Comparison of Soil Microbial Communities Between Artificial Tea Plantation and Wild Tea Plantation in China
ABSTRACT
Soil microorganisms can strengthen the material and energy cycle in the ecosystem and are an important indicator of the stability of the soil ecosystem. Soil microorganisms in tea plantations include bacteria, actinomycetes, and fungi. Bacteria are the main player in terms of quantity and play an important role in the soil ecology of tea plantations. This study uses high-throughput sequencing technology to analyze the ecological diversity of bacterial communities in Lingyun Baihao tea plantations in Guangxi, analyzes the composition of soil bacterial communities in 3 different types of tea plantations in Lingyun County, Guangxi, and compares the differences in bacterial communities between them. To promote the understanding of the soil micro-ecology of Guangxi Lingyun Baihao tea plantation. Our research confirmed the diversity and composition of soil microbial communities in Guangxi Lingyun Baihao tea garden. Among them, Proteobacteria, Acidobacteria, Bacteroidetes and Actinobacteria are the dominant communities in the tea garden ecosystem, revealing that the intensity of human agricultural production may affect the diversity of the bacterial community in tea gardens. Their structure and function provide a reference for understanding the diversity of soil microbial community in Lingyun Baihao tea garden in Guangxi.
KEYWORDS
Artificial tea plantation; Wild tea plantation; Soil microbial communities; High-throughput sequencing technology
INTRODUCTION
Plants exert a strong influence on the composition of microbial communities in soil through rhizodeposition and the decay of litter and roots. Consequently, the study of forest microbial communities and their relationship with tea plantation is of paramount importance for understanding the ecosystem services provided by soil microorganisms. A better understanding of this could allow us to confer an “economic value” to native habitats, there by quantitatively promote their conservation. Biological control agents along with plant growth promoting and other important antimicrobial communities can achieve plant-growth promoting and stress-ameliorating capabilities, as well as suppressing soilborne pathogens.
Soil microbiota is critical in establishing healthy soil. The soil inhibition is a plant defense mechanism established by natural microorganisms in soil, which provides the first defense barrier for soil-borne plant pathogens [1,2]. Industrial agriculture is directly beneficial to improving labor efficiency and crop production. Intense fertilizer use has led to a series of ecological environmental problems such as loss of diversity in ecosystems, decreases in soil fertility and aggravation of environmental pollution [3,4]. Soil microorganisms account for a large part of the earth’s biodiversity and play a significant role in soil ecosystem biochemical processes, such as nutrient cycling and soil-borne pathogen suppression [5].
Any disturbance from both natural and anthropogenic origin may cause changes in microbial communities. Many studies are focused on the description of the variations in microbial community composition and their correlation to functional roles [6-10]. Microbial role in the environment can be seriously altered. In short term, the farmers are able to obtain high crop yields by using pesticides and chemical fertilizers. They usually affecting soil quality, microbial activities and populations. Based on the hypothesis that native microbiota composition represents a soil quality indicator, the timely detection of microbial community alterations is crucial for the development of sustainable tea plantation practices.
In this study, Tea trees soil were used to investigate the effect on the rhizosphere bacterial community. Dynamic succession of the bacterial community structure in the rhizosphere was explored by 16S rDNA gene high-throughput sequencing. These results provide a comprehensive understanding of the rhizosphere microbial response to tea tree influence and may help improve farming strategies by regulating the structure and function of rhizosphere soil flora to reduce the negative impacts. In view of the importance of soil microbial communities to healthy soil ecosystems, this study used high-throughput sequencing technology to analyze the ecological diversity of soil bacterial communities in Guangxi Lingyun Baihao tea plantations with different levels of human agricultural production activities and analyzed the composition of three different types of soil bacterial community. The differences in bacterial communities in tea gardens in Lingyun County, Guangxi were compared. The impact of human agricultural production activities on the soil bacterial communities in tea plantations was assessed to enhance the understanding of soil micro-ecology in Lingyun Baihao tea plantations in Guangxi.
MATERIALS AND METHODS
Collection and DNA Extraction of Soil Sample
The soil samples were collected from Baihao tea plantations in Lingyun County, Baise City, Guangxi, China, which are state-owned tea factory (GYCC) artificial ancient tea plantation (N:24°31´43”, E: 106°32´35”, Langfu(F) artificial organic tea plantations(N: 24°46´19”, E: 106°58´60”) and Xianfengling(XFL) wild tea plantation (N: 24°32´4”, E: 106°32´19”).
DNA Extraction and 16S rDNA Sequencing
DNA was extracted from 0.25g of each soil sample using the Power Soil DNA isolation kit (MoBio Laboratories Inc., Carlsbad, CA, USA) following the manufacturer’s instructions. The extracted DNA samples were analyzed using a Nanodrop 2000 UV-Vis spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and the DNA quality was confirmed by 1% agarose gel electrophoresis. After the total DNA of the sample is extracted, specific primers with Barcode are synthesized according to the full-length primer sequence, PCR amplification is performed, and the products are purified, quantified, and homogenized to form a sequencing library (SMRT Bell). The built library is first subjected to library quality Check, the library qualified by the quality check is sequenced with PacBio Sequel. The PacBio Sequel off-machine data is in bam format, and the CCS file is exported through smart link analysis software, and the data of different samples are identified according to the barcode sequence and converted into fastq format data.
Diversity Analysis of Soil Microbial Community
After exporting the PacBio off-machine data to a CCS file (the CCS sequence is obtained using the smart link tool provided by Pacbio), there are mainly three steps as follows: The first step is the CCS identification using lima v1.7.0 software which identifies the CCS through the barcode, obtained Raw-CCS sequence data. The second step is CCS filtering. Use cutadapt 1.9.1 software to identify and remove primer sequences and perform length filtering to obtain Clean-CCS sequences that do not contain primer sequences. The third step is to remove chimeras: use UCHIME v4.2 software to identify and remove After removing the chimera sequence. Following these three steps, the effective-CCS sequence is obtained. Use arch software to cluster Reads at a similarity level of 97.0% to obtain OTUs. Take SILVA as the reference database and use the Naive Bayes classifier combined with the method of comparison to perform taxonomic annotations on the feature sequence. Then the species classification information corresponding to each feature can be obtained. Then at each level (phylum, class, order, family, genus, species) count the community composition of each sample. Use QIIME software to generate species abundance tables at different taxonomic levels. Then use R language tools to draw community structure diagrams at each taxonomic level of the samples. Use QIIME2 software to carry out the sample Alpha diversity index and Beta diversity index analysis. Use Mother software and R language tools to draw a Shannon diversity index dilution curve based on the Shannon index of each sample’s sequencing amount at different sequencing depths to reflect the microbial diversity of each sample at different sequencing quantities. Use PICRUSt2 software to use the feature sequence for prediction and the existing phylogenetic tree in the software for species annotations. Use IMG microbial genome data to output functional information and infer the functional gene composition in the sample. Thereby assess different samples or groupings for functional difference.
RESULTS
Evaluation of Soil Sample Sequencing Data Quality
Among the three tea plantations in this study, XFL tea plantations have the least artificial agricultural production activities, followed by GYCC tea plantations, and LF tea plantations have the most intense artificial agricultural production activities. The tea trees in LF tea plantations grew poorly and caught a lot of parasitic fungi; the tea trees in XFL and GYCC tea plantations grew well without parasitic fungi Figure 1.
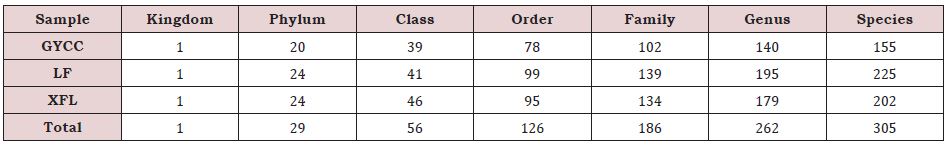
PacBio Sequel was used to sequence and analyze the bacterial community composition of the three mixed soil samples. The sequencing data evaluation results of each sample are shown in Table 1. After filtering, a total of 20757 high-quality sequences were obtained, with an average sequence length of 1451bp (Table 1). Rarefaction Curve is to randomly select a certain number of sequences from a sample, count the number of species represented by these sequences, and construct a curve based on the number of sequences and the number of species to verify whether the amount of sequencing data is sufficient to reflect the species diversity in the sample. The Rarefaction Curve shows that the slope of the curve of most samples has been significantly reduced, and there is a trend toward saturation, which to a certain extent reflects that the depth of sequencing in this experiment met the needs of analysis at a level above. Sample ID is the name of the tea plantation sample; Raw- CCS is the number of CCS identified by the sample; Clean CCS is the number of sequences after primer removal and length filtering; Effective-CCS is the number of sequences used for subsequent analysis after the chimera is removed; Avg Len (bp) is the average sequence length of the sample; Effective (%) is the percentage of Effective-CCS in Raw-CCS.
Analysis of OTUs in Soil samples from Tea Plantations
In bioinformatics data analysis, each sequence obtained by sequencing comes from a corresponding strain. Through the classification operation, the sequences are classified as Operational Taxonomic Units (OTU) according to the similarity between the sequences. When divided according to 95% similarity, an OTU is a collection unit of sequences belonging to the same genus; when divided according to 97% similarity, an OTU is classified as a sequence collection of one species. In order to analyze the bacterial community structure of tea plantation soil, Usearch software was used to cluster reads at a similarity level of 97.0%. Total 899 OTUs were obtained from three mixed soil samples (Figure 2-4). Among them, LF tea plantation soil has the most OTUs: LF tea plantation soil contains 553 OTUs, GYCC tea plantation soil contains 403 OTUs, and XFL tea plantation soil contains 488 OTUs (Figure 2). Comparing the number of communities shared between the three tea plantations soil samples (Figure 2) there are 96 OTUs shared by the soil of the three tea plantations. Among them, GYCC and LF tea plantations have 171 identical OTUs. GYCC and XFL tea plantations have 13 identical OTUs. LF and XFL tea plantations share 169 OTUs. In addition, there are plantation specific communities in the soil of these three tea plantations: The GYCC tea plantation has 123 plantations specific OTUs, the LF tea plantation has 117 plantation specific OTUs, and the XFL tea plantation has 210 plantation specific OTUs. The samples of different tea plantations are represented by different colors. The number of overlapping parts between multiple color graphs is the number of features shared by multiple samples, and the non-overlapping part is the number of unique features of each sample.
α-Diversity Index Analysis of Soil Bacterial Community Diversity in Tea Plantations
Alpha diversity reflects the species abundance and species diversity of a single sample. There are multiple measurement indicators. The Chao1 and Ace indexes measure species abundance. The Shannon and Simpson indexes are used to measure species diversity and are affected by sample Community evenness. In the case of the same species abundance, the greater the uniformity of each species in the community, the greater the diversity of the community. The greater the Shannon index and Simpson index, the higher the species diversity of the sample.
The Coverage indexes of the three tea plantations soils are all greater than 0.98 indicating that the species in the samples have a high probability of being detected, and once again that the sequencing results better reflect the community structure and species diversity in the tea plantation soil. For Chao1 and Ace indexes, both are in following order: LF>XFL>GYXC. That is, LF tea plantation has the largest number of communities while GYCC tea plantation has the least number. This is consistent with the OTUs community analysis result. For the Simpson and Shannon indices, both are LF>GYCC>XFL, indicating that for the same species abundance, LF tea plantation has the highest species diversity, GYCC tea plantation is the second, and XFL tea plantation is the lowest (Table 2).
Sample ID is the name of the sample; Feature is the number of features (OTUs); Chao1, Ace, Shannon, and Simpson respectively represent each index; Coverage is the coverage rate of the sample library.
Analysis of bacterial community structure in the soil of tea plantation
At each classification level of bacteria (phyla, class, order, family, genus, species), the composition of bacterial communities in the soil of three tea plantations was counted, and the results are shown in the (Table 3). The results show that the soil of LF tea Plantation has the most types of bacteria at all classification levels (except for the family level which is slightly lower than XFL tea plantation), followed by XFL tea plantation, and GYCC tea plantation have the least, which is the same as OTUs analysis.
At the level of phylum classification, the composition of the soil bacterial community in three tea plantations was analyzed. The most dominant bacterial communities in the three tea plantation soils are all Proteobacteria. The Proteobacteria community in the XFL tea plantation soil has the most obvious community advantages. The second most dominant bacterial group is Acidobacteria. Comparing the differences in dominance of Acidobacteria among the three tea plantations, the order in dominance of Acidobacteria in the soil is GYCC>LF>XFL. Verrucomicrobia is the third most dominant community. It has a higher predominance in the soil of LF tea plantations than GYCC and XFL. In addition, compared with GYCC and XFL, LF tea plantation soil has a higher relative abundance advantage of Bacteroidetes and Bacteroidota bacterial communities. It is worth noting that the relative abundance advantage of the bacterial communities Gemmatimonadetes and Rokubacteria in XFL tea plantation soil is high while the relative abundance advantage in GYCC and LF tea plantation soil is extremely low. For the bacterial community Acidobacteriota, it also occupies a certain relative abundance in the soil of GYCC and LF tea plantations. However, it is not found in the soil of XFL tea plantations. There are significant differences in the relative abundance of bacterial communities in the soil of the three tea gardens at the phylum level (Figure 3).
The soil bacterial communities of the three tea plantations were also analyzed at the genus level. Most of the dominant bacterial communities of the three tea plantations were uncultrured bacterium. There are only relatively few genera that can be cultivated: Candidatus Udaeobacter, Sphingomonas, Candidatus Solibacter, and ADurb.Bin063-1. The most abundant cultivable bacteria in the soil of XFL tea plantations are Candidatus Udaeobacter and Sphingomonas. The genus of culturable bacteria with higher abundance in the soil of LF tea plantation is ADurb. Bin063-1. The genus of culturable bacteria with higher abundance in the soil of GYCC tea plantation is Candidatus Solibacter (Figure 4).
A color represents a species, and the length of the color block represents the relative abundance proportion of the species. To make the view the best, only display the top ten species with abundance level, and merge the other species into others to display in the figure. The corresponding value of the heat map is the Z value obtained after the relative abundance of each row of species is standardized. The color gradient from blue to red indicates the relative abundance from low to high; the previous row in the figure is the sample grouping information, the color and the graph Column correspondence.
Analysis On The Differences Of Soil Bacterial Communities In Three Tea Plantations
The ternary phase diagram uses an equilateral triangle to describe the ratio relationship of the different attributes of the three variables. In this analysis, the species composition of three or three groups of samples can be compared and analyzed according to the species classification information. The triangle diagram can be intuitively displayed and draw out the proportions and relationships of different species in the sample. The Ternary Phase Diagram of soil bacterial communities in three tea plantations (Figure 5).
The three corners of the triangle represent three tea plantations, which are represented by three colors. The three sides are used to measure the abundance of the corresponding tea plantations. The icons in the triangle represent the number of all species contained in a certain level. The size of the icon represents the average relative abundance of the species, and the colored icons in the legend represent the species classifications of the five phylum levels with the highest abundance. At the phylum level, The top five bacterial communities in relative abundance were listed separately, and the rest were combined into others. The differences of bacterial communities among the three tea plantations were analyzed. The types of high relative abundance of bacterial communities in XFL tea plantation soil are Proteobacteria and other types. Compared with GYCC tea plantation and LF tea plantation, the high relative abundance of other types of bacterial communities in XFL tea plantation soil is more obvious. The relative abundance of Proteobacteria and Planctonycetes in the soil of GYCC tea plantations is larger. Compared with XFL tea plantation and LF tea plantation, its higher relative abundance of Planctonycetes is more obvious. For LF tea plantation, the relative abundance of Verrucomicrobia is greater than that of GYCC tea plantation and LF tea plantation.
Functional Prediction of Soil Microbial Communities
In order to better study the micro-ecological functions of soil microorganisms in the three tea plantations, we used PICRUSt2 software for Clusters of Orthologous Groups of proteins (COG) analysis. The results showed that the functional characteristics of the soil bacterial communities in the three tea plantations were significantly different. The metabolic functions of the soil bacterial communities were rich, most of which belonged to the metabolism, and the bacterial communities were metabolically active. Comparing the differences in the metabolic pathways between LF tea plantations and XFL tea plantations, most of the metabolic pathways in XFL tea plantations are stronger than LF tea plantations. These metabolic pathways include Coenzyme transport and metabolism; Energy production and conversion; Translation, ribosomal structure, and biogenesis; Amino acid transport and metabolism; Cell wall/ membrane/envelope biogenesis. For the two metabolic pathways of Carbohydrate transport and metabolism and Transcription, LF tea plantations have big advantages. Comparing the differences in the metabolic pathways between LF tea plantations and GYCC tea plantations, the strength of the Coenzyme transport and metabolism; Translation, ribosomal structure and biogenesis; Amino acid transport and metabolism pathways of GYCC tea plantations is greater than that of LF tea plantations. The Inorganic ion transport and metabolism pathway of LF tea plantation is stronger than that of GYCC tea plantation. For Energy production and conversion and Carbohydrate transport and metabolism; Transcription; Cell wall/membrane/envelope biogenesis pathways, there is little difference between LF tea plantations and GYCC tea plantations. The results of function prediction also reflect that artificial agricultural production activities have a significant impact on the function of soil bacterial communities in tea plantations, especially Energy production and conversion and Carbohydrate transport and metabolism; Transcription; Cell wall/membrane/ envelope biogenesis (Figure 6).
The abscissa is the relative abundance percentage of the species’ metabolic pathway, and the ordinate is the name of the metabolic pathway. The different colors correspond to the three tea plantation samples. The left figure shows the abundance ratio of different functions in two samples or two sets of samples, the middle shows the difference ratio of the function abundance within the 95% confidence interval, and the rightmost value is the p value.
DISCUSSION
Tea trees originated in the Yunnan-Guizhou Plateau in southwestern China. Guangxi is one of the main tea producing areas in China with a long history of tea development. The Lingyun Baihao tea is rich in substance. Its tea soup has a strong taste, strong aroma and unique quality. It is one of the most important local tea varieties in Guangxi. As the largest artificial ecosystem on the planet, the issue of biodiversity in agricultural ecosystems has attracted more and more attention from academic circles and even mankind. Due to serious human interference, its biodiversity often fails to meet the requirements of a natural and stable ecology. This study uses high-throughput sequencing technology to analyze the ecological diversity of bacterial communities in Lingyun Baihao tea plantations in Guangxi, analyzes the composition of soil bacterial communities in three different types of tea plantations in Lingyun County, Guangxi, and compares the differences in bacterial communities between them. In order to promote the understanding of the soil micro-ecology of Guangxi Lingyun Baihao tea plantation.
Soil microorganisms are one of the important components of the soil ecosystem. They can strengthen the material and energy cycle in the ecosystem and are an important indicator of the stability of the soil ecosystem. Soil microorganisms in tea plantations include bacteria, actinomycetes, and fungi. They are mainly bacteria in quantity, and they are an important part of life in the soil ecology of tea plantations [11,12]. Existing studies have shown that land use has a significant impact on soil bacterial communities [13,14]. Soil microorganisms in tea plantations are also affected by environmental conditions and agricultural activities [15]. Therefore, studying the abundance, composition and diversity of bacterial communities in the soils of different plantations of Guangxi Lingyun Baihao Tea is essential for the sustainable management and production of Guangxi Lingyun Baihao Tea. Through the OTUs analysis of the bacterial communities in the soil of different tea plantations, it was found that the soils of the three tea plantations shared 96 species, accounting for about 10.68% of the total OTUs, indicating that environmental conditions and agricultural activities have a greater impact on soil Bacterial community of tea plantings. LF tea plantation has the largest population, as high as 553 OTU, which may be related to the frequent agricultural production activities of humans, because compared with XFL and GYCC tea plantations, the agricultural production activities of LF tea plantations are more frequent and the degree of human intervention is greater. Thus, the number of bacterial OTUs has risen. However, the XFL tea plantation without human intervention has a higher OTUs than the GYCC tea plantation with relatively little human production intervention. This again shows that human agricultural production activities will affect the diversity of the soil bacterial community in the tea plantation. The impact mechanism is not yet clear. According to the analysis of alpha diversity of soil bacterial communities in different tea gardens, the results of all indexes (including Chao1, Ace, Simpson and Shannon indexes) are LF>XFL>GYXC, indicating that LF has the largest number of bacterial communities and GYCC tea plantation has the lowest. The results are consistent with OTUs analysis. It shows that compared with XFL tea plantation, the soil species diversity of the two are evolutionarily closer, and it also shows that artificial agricultural production affects the microecological changes of the agricultural ecosystem. Ederson et al. reported that the use of land by humans has changed the structure of the soil bacterial community in the Western Amazon. After the introduction of crops or pastures, the structure of the soil bacterial community has changed. But the intensity of slash-and-burn agriculture is relatively weak and the impact is not particularly significant [16]. Lynn et al. reported that land use has a great influence on the abundance, diversity and community composition of soil microorganisms [17]. The difference between the results of this research and Ederson’s research also shows that modern agricultural production activities in LF and GYCC tea plantations are more intense than primitive slash-and-burn agriculture.
The diversity of soil bacterial communities is of great significance to maintaining the stability and sustainability of soil ecosystems [18]. The studies have shown that in the soil of agricultural systems, the most common bacterial communities are Proteobacteria, Chloroflexi, Acidobacteria, Actinobacteria and Bacteroidetes [19,20]. In the study of bacterial community diversity in the soil ecosystem of tea plantations, Zhang et al. reported that Proteobacteria, Acidobacteria, Bacteroidetes and Actinobacteria are the main bacterial communities in the soil ecosystem of tea plantations [21]. The research data of Ji et al. also reported similar results [21]. This study analyzed the differences in the soil bacterial community structure of the tea plantations in the three habitats of LF tea plantation, GYCC tea plantation and XFL tea plantation from the taxonomic level of phylum and genus. Proteobacteria is the most common bacterial group in each of the three tea plantations. Acidobacteria, Bacteroidetes and Actinobacteria also exist in the soil of these three tea plantations.This is consistent with the results of previous studies.
At the phylum level, there are significant differences in the composition of soil bacterial communities in three different tea plantations. The top two bacterial communities in relative abundance are Proteobacteria and Acidobacteriota. Comparing the differences in soil bacterial communities among GYCC tea plantation, LF tea plantation and XFL tea plantation, the relative abundance of Proteobacteria and other types of bacterial communities in the soil of XFL tea plantation is higher, compared to the other two tea plantations. The relative abundance advantage of the population is more significant. Among other types of bacterial communities in the soil of XFL tea plantation, Rokubacteria and Gemmatimonadetes have a higher abundance. Compared with other soils, GYCC tea plantation has a more significant advantage in relative abundance of Planctonycetes. For LF tea plantations, the relative abundance of Verrucomicrobia is more significant. Studies have shown that Proteobacteria and Acidobacteria are two important phylums in the microbial community. They have the ability to degrade cellulose and lignin in plant residues and play an important role in the carbon cycle. They can enhance the circulation of essential nutrients and improve the soil Fertility and sustainability [ 23-26]. However, the community functions of Proteobacteria and Acidobacteria are also different. Proteobacteria adapts to resource-rich environments while Acidobacteria are just the opposite [27-29]. Ji et al. reported that the organic substitution rate of tea plantations was positively correlated with the relative abundance of Proteobacteria, but negatively correlated with the relative abundance of Acidobacteria [21,22]. Proteobacteria, the important phyla in the microbial community, may use unstable carbon sources and grow rapidly in a nutrient-rich environment, forming a relatively high relative abundance [27,30]. Zhang et al. reported that the soil microbial community structure in organically covered tea plantations. They found that the Proteobacteria and Acidobacteria in the soil covered by peanut shells were higher than those covered by polyethylene film. It shows that peanut shells as crop residues can provide rich nutrition for Proteobacteria and Acidobacteria [31]. Gemmatimodetes and Rokubacteria have been shown to have multiple pathways encoding antibacterial secondary metabolites [32,33]. Planctonycetes are widely distributed in natural systems such as soil. They can use ammonium nitrogen (NH4+) and nitrite nitrogen (NO2−) to generate nitrogen in anoxic environment to obtain energy and regulate the nitrogen source of the farmland soil system with excessive application of nitrogen fertilizer. It is of great significance to the nitrogen cycle of the soil [34-36]. Verrucomicrobia has the ability to utilize carbon sources such as cellulose [37]. Herlemann et al. found that Verrucomicrobia plays an important role in the carbon cycle of marine ecosystems [38]. In addition, Khadem et al. also reported that members of Verrucomicrobia are involved in nitrogen fixation in the soil [39]. At the genus level, the majority of soil bacterial communities are uncultured bacterium. There are relatively few cultivable bacteria. The cultivable bacterial genera in the soil of the three tea plantations of XFL, GYCC and LF are Candidatus Udaeobacter and Sphingomonas, Candidatus Solibacter and ADurb.Bin063-1, respectively.
The microecological function analysis results of soil microorganisms show that most of the metabolism in soil samples belongs to metabolism (General function prediction only,Amino acid transport and metabolism, Translation,ribosomal structure and biogenesis, Cell wall/membrane/envelope biogenesis, Function unknown, Energy production and conversion, Transcription, Carbohydrate transport and metabolism; Coenzyme transport and metabolism, Inorganic ion transport and metabolism, etc.).The bacterial metabolism is active. The results of this research are similar to those previously published [31,40]. In addition to the General function prediction only metabolic pathway, the Amino acid transport and metabolism metabolic pathway is the most active, which also shows that the nitrogen cycle in the tea plantation soil is very strong. For XFL tea plantation, Energy production and conversion, Translation, ribosomal structure and biogenesis and Coenzyme transport and metabolism are more active than GYCC tea plantation and LF tea plantation. This shows that the carbon cycle in the soil of XFL tea plantation is stronger than those of GYCC tea plantation and LF tea plantation. This result is consistent with the analysis result of bacterial community composition. Comparing the differences in metabolic pathways among LF tea plantations and XFL and GYCC tea plantations, most of the metabolic pathways in XFL tea plantations are stronger than those in LF tea plantations. GYCC tea plantations have stronger metabolic pathways than LF tea plantations and XFL tea plantations. It is worth noting that for Energy production and conversion and Carbohydrate transport and metabolism; Transcription; Cell wall/membrane/ envelope biogenesis metabolic pathways, XFL tea plantations are stronger than LF tea plantations, while GYCC tea plantations and LF tea plantations are basically Close. This may be because with the deepening of human agricultural production activities, the types of soil bacterial communities have increased, and there has been fierce competition between nutrients and living space, which in turn affects the distribution of metabolic flux. In summary, the diversity and composition of soil bacterial communities in tea plantations play a very important role in the circulation of soil nutrients. Establishing a microbial early warning and regulation system for the virtuous cycle of the tea plantation soil ecosystem, guiding the scientific management of the tea plantation soil, and protecting the ecological environment of the tea plantation are of important practical significance for promoting the sustainable and healthy development of tea production.
CONCLUSION
In view of the importance of soil microbial communities to healthy soil ecosystems, our study clarified the diversity and composition of soil microbial communities in Guangxi Lingyun Baihao tea plantation. The research results showed that Proteobacteria, Acidobacteria, Bacteroidetes and Actinobacteria are the dominant communities in the tea plantation ecosystem. It also revealed that the intensity of human agricultural production may affect the structure and function of the bacterial community diversity in tea plantations and provided a reference for understanding the diversity of soil microbial communities in Guangxi Lingyun Baihao tea plantation.
AUTHOR CONTRIBUTIONS
Liqin Zhou designed the experiments. Xiaohu Wang and Liujian Ye performed most of the experiments and wrote the manuscript.
FUNDING
This study was financially supported by the Science and Technology Project of Guangxi (2020ZYZX3027, AB2107601) funding from the Bagui Scholar Program Fund (2016A25) of Guangxi Zhuang Autonomous Region provided to LQ.Z.
REFERENCES
- Trivedi P, Leach JE, Tringe SG (2020) Plant microbiome interactions from community assembly to plant health. Nat Rev Microbiol 18(11): 607-621.
- Mendes R, Kruijt M, de Bruijn I (2011) Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332(6033): 1097- 1100.
- Gomes NCM, Fagbola O, Costa R (2003) Dynamics of fungal communities in Bulk and maize rhizosphere soil in the tropics. Applied and Environmental Microbiology 69(9): 5736.
- Jacobsen S, Sørensen M, Pedersen SM (2013) Feeding the world genetically modified crops versus agricultural biodiversity. Agronomy for Sustainable Development 33(4): 651-66.2.
- Zhang K, Cheng X, Shu X (2018) Linking soil bacterial and fungal communities to vegetation succession following agricultural abandonment. Plant and Soil 431(1): 19-36.
- Fierer N, Ladau J, Clemente JC (2013) Reconstructing the microbial diversity and function of pre-agricultural Tallgrass prairie soils in the United States. Science 342(6158): 621-624.
- Fierer N, Lauber CL, Ramirez KS (2012) Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. The ISME Journal6(5): 1007-1017.
- Griffiths RI, Thomson BC, James P (2011) The bacterial biogeography of British soils. Environmental Microbiology13(6): 1642-1654.
- Wu T, Chellemi DO, Graham JH (2008) Comparison of Soil Bacterial Communities under Diverse Agricultural Land Management and Crop Production Practices. Microbial Ecology 55(2): 293-310.
- Mendes LW, Tsai SM, Navarrete AA (2015) Soil-borne microbiome: linking diversity to function. Microbial Ecology.
- Fierer N (2017) Embracing the unknown: disentangling the complexities of the soil microbiome. Nature Review Microbiology 15(10): 579-590.
- Sun S, Li S, Avera BN (2017) Soil bacterial and fungal communities show distinct recovery patterns during forest ecosystem restoration. Appl Environ Microbiol 83(14).
- Bissett A, Richardson AE, Baker G (2011) Long-term land use effects on soil microbial community structure and function. Applied soil ecology: a section of Agriculture, ecosystems and environment 51: 66-78.
- Liliensiek A, Thakuria D, Clipson N (2012) Influences of plant species composition, fertilization and lolium perenne ingression on soil microbial community structure in three irish grasslands. Microbial Ecology 63(3): 509-521.
- Han W, Xu J, Wei K (2013) Soil carbon sequestration, plant nutrients and biological activities affected by organic farming system in tea (Camellia sinensis (L) O Kuntze) fields. Soil science and plant nutrition 59(5): 727- 739.
- Ederson DJ, Marsh TL, Tiedje JM (2009) Changes in land use alter the structure of bacterial communities in Western Amazon soils. The ISME Journal 3(10): 1222.
- Lynn TM, Liu Q, Hu Y (2017) Influence of land use on bacterial and archaeal diversity and community structures in three natural ecosystems and one agricultural soil. Arch Microbiol 199(5): 711-721.
- Zhao J, Ni T, Li Y (2014) Responses of bacterial communities in arable soils in a rice-wheat cropping system to different fertilizer regimes and sampling times. PloS One 9(1): e85301.
- Yang Y, Li X, Liu J (2017) Bacterial diversity as affected by application of manure in red soils of subtropical China. Biology and fertility of soils 53(6): 639-649.
- Tian W, Wang L, Li Y (2015) Responses of microbial activity, abundance, and community in wheat soil after three years of heavy fertilization with manure-based compost and inorganic nitrogen. Agriculture, Ecosystems and environment 213: 219-227.
- Zhang S, Sun L, Wang Y (2020) Cow manure application effectively regulates the soil bacterial community in tea plantation. BMC microbiology 20(1): 190.
- Ji L, Wu Z, You Z (2018) Effects of organic substitution for synthetic N fertilizer on soil bacterial diversity and community composition: A 10-year field trial in a tea plantation. Agriculture, Ecosystems and Environment 268: 124-132.
- Eichorst SA, Trojan D, Roux S (2018) Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environmental Microbiology 20(3): 1041-1063.
- Ward NL, Challacombe JF, Janssen PH (2009) Three Genomes from the phylum acidobacteria provide insight into the lifestyles of these microorganisms in soils. Applied and Environmental Microbiology 75(7): 2046-2056.
- Randall TE, Fernandez Bayo JD, Harrold DR (2020) Changes of Fusarium oxysporum f.sp. lactucae levels and soil microbial community during soil biosolarization using chitin as soil amendment. PloS One 15(5): e232662.
- Huang X, Liu L, Wen T (2015) Illumina MiSeq investigations on the changes of microbial community in the Fusarium oxysporum f.sp. cubense infected soil during and after reductive soil disinfestation. Microbiological Research 181: 33-42.
- Kuramae EE, Yergeau E, Wong L (2012) Soil characteristics more strongly influence soil bacterial communities than land-use type. FEMS Microbiology Ecology 79(1): 12-24.
- Inceoglu O, Abu Al Soud W, Salles JF (2011) Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing. PloS One 6(8): e23321.
- Janssen ph (2006) Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Applied and environmental microbiology 72(3): 1719-1728.
- Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology (Durham) 88(6): 1354-1364.
- Zhang S, Wang Y, Sun L (2020) Organic mulching positively regulates the soil microbial communities and ecosystem functions in tea plantation. BMC microbiology 20(1): 103.
- Peterson SB, Bertolli SK, Mougous JD (2020) The central role of interbacterial antagonism in bacterial life. Current Biology 30(19): R1203-R1214.
- Crits Christoph A, Diamond S, Butterfield CN (2018) Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature (London) 558(7710): 440-444.
- Ivanova AO, Dedysh SN (2012) Abundance, diversity and depth distribution of planctomycetes in acidic northern wetlands. Frontiers in Microbiology 3: 5.
- Nie SA, Lei X, Zhao L (2018) Response of activity, abundance and composition of anammox bacterial community to different fertilization in a paddy soil. Biology and fertility of soils 54(8): 977-984.
- De Cocker P, Bessiere Y, Hernandez-Raquet G (2018) Enrichment and adaptation yield high anammox conversion rates under low temperatures. Bioresource Technology 250: 505-512.
- Dunfield PF, Yurgey AQ, Senin P (2007) Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature (London) 450(7171): 879-882.
- Herlemann DPR, Lundin D, Labrenz M (2013) Metagenomic de novo assembly of an aquatic representative of the verrucomicrobial class Spartobacteria. MBio 4(3): e512-e569.
- Khadem AF, Pol A, Jetten MS (2010) M Nitrogen fixation by the verrucomicrobial methanotroph ‘methylacidiphilum fumariolicum’ solv. Microbiology 156: 1052-1059.
- Zhang Y, Hu A, Zhou J (2020) Comparison of bacterial communities in soil samples with and without tomato bacterial wilt caused by Ralstonia solanacearum species complex. BMC Microbiology 20(1): 89.
Article Type
Research Article
Publication history
Received Date: August 17, 2022
Published: August 30, 2022
Address for correspondence
Liqin Zhou, Key Laboratory of Non-Food Biomass and Enzyme Technology, Guangxi Academy of Sciences, China
Copyright
©2022 Open Access Journal of Biomedical Science, All rights reserved. No part of this content may be reproduced or transmitted in any form or by any means as per the standard guidelines of fair use. Open Access Journal of Biomedical Science is licensed under a Creative Commons Attribution 4.0 International License
How to cite this article
Xiaohu W, Liujian Y, Shengbo W, Shuang H, Qixia Z, Liqin Z. Comparison of Soil Microbial Communities Between Artificial Tea Plantation and Wild Tea Plantation in China. 2022- 4(4) OAJBS.ID.000480.
Figure 1: Tea tree diagrams of the three tea plantations.
LF: Langfu artificial organic tea plantation in Lingyun County, Baise City, Guangxi; GYCC: the artificial ancient tea
plantation of the State-owned tea plantation in Lingyun County,Baise City, Guangxi; XFL: the Xianfengling wild tea
plantation in Lingyun County, Baise City, Guangxi.

Figure 2: Venn diagrams of soil microbial communities in three tea plantations.
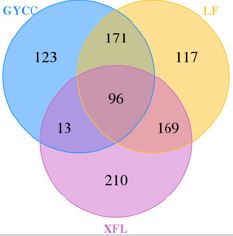
Figure 3: Relative abundance of soil phylum bacterial communities in three tea plantations.
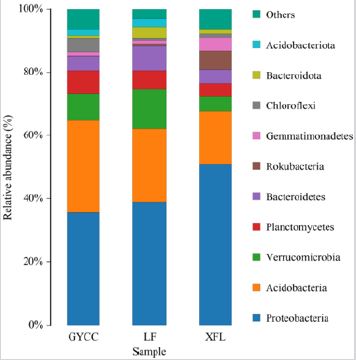
Figure 4: Clustering heat map of soil genus abundance of bacterial communities in three tea plantations.
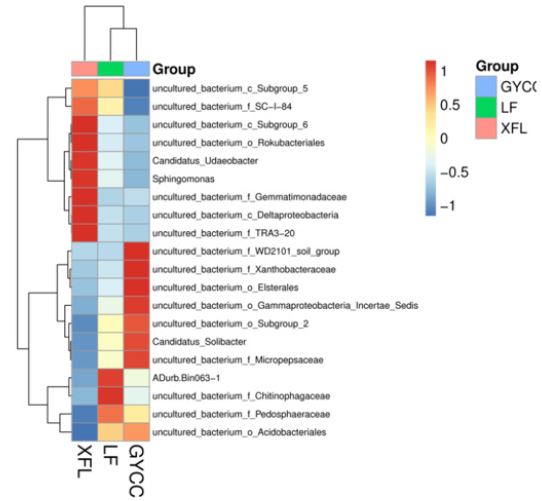
Figure 5: The ternary phase diagram of the differences in soil bacterial communities in three tea plantations.
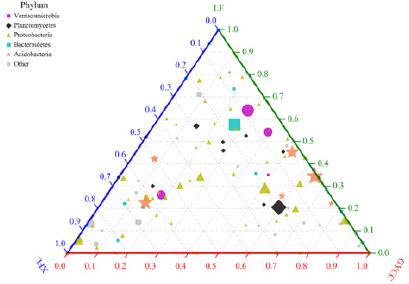
Figure 6: The COG functional classification statistics map of three tea plantation soils.
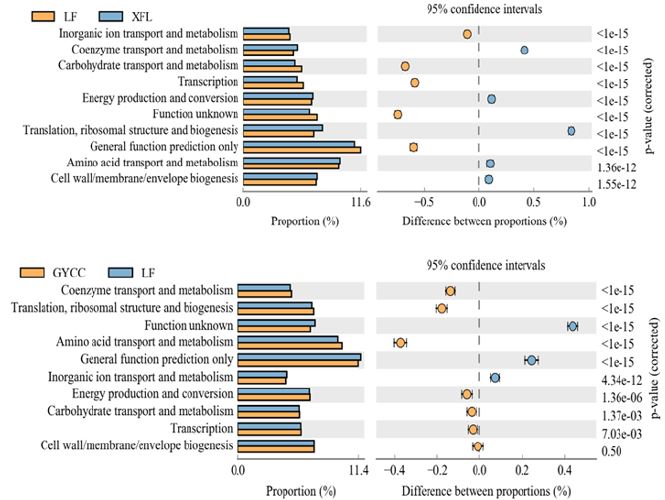
Table 1: Statistics of the three tea plantations sequencing data processing results.
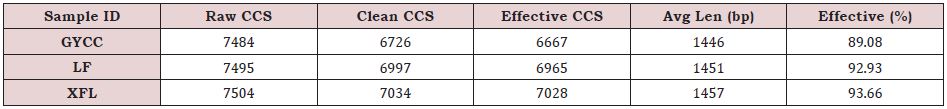
Table 2: Statistics of α-diversity index of bacterial communities in the soil of tea plantations.
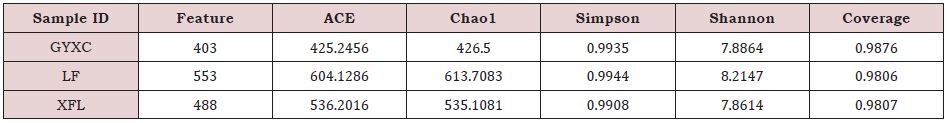
Table 3: The statistical table of species of various grades of bacterial communities in the soil of three tea plantations.