Molecular Mimetism Between Cystic Fibrosis and P. Aeruginosa and S. Aureus, Possible Cause of Autoimmunity? In-Silico Analysis
ABSTRACT
Background: Cystic fibrosis is mainly caused by a genetic mutation, through a gene located on chromosome 7. The result of
all mutations is decreased chloride secretion and, consequently, increased reabsorption of sodium in the cell space. The disease
occurs as a cascade effect following infection and the subsequent inflammatory process. The most common bacteria that cause
endobronchial infection are Staphylococcus aureus and Pseudomonas aeruginosa.
Methodology: Amino acid sequences of autoantigens for cystic fibrosis were selected from the AAgAtlas autoantigen database
(http://biokb.ncpsb.org/aagatlas/). The amino acid sequence of each autoantigen was used as input in PSI-BLAST (https://blast.
ncbi.nlm.nih.gov/Blast.cgi) to find similar antigens of pathogens associated with infection in cystic fibrosis. For epitope prediction,
the autoantigens analyzed in this study were used as entry for the Ellipro server (http://tools.iedb.org/ellipro/).
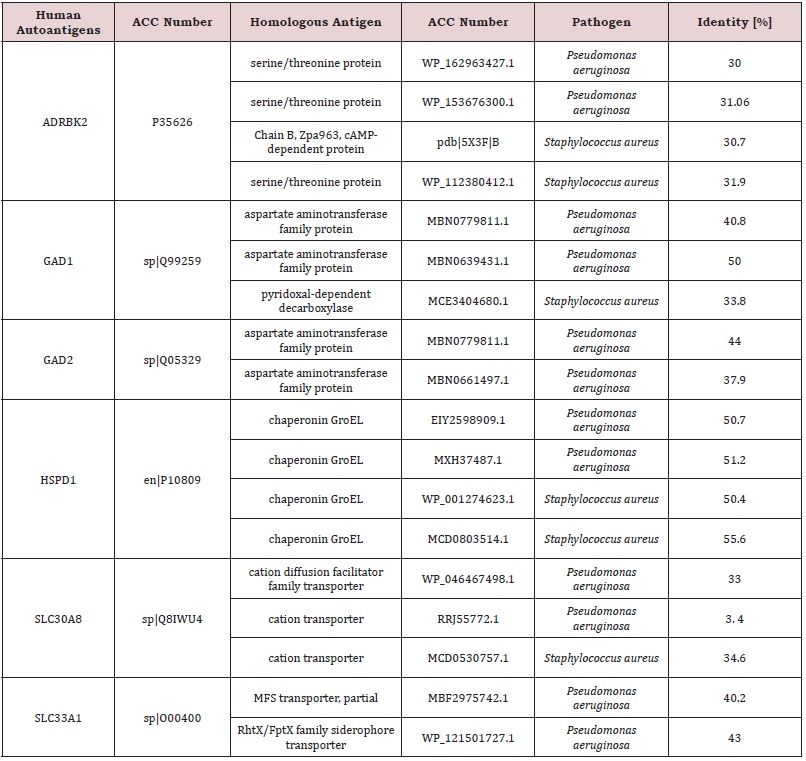
Results: Autoantigens ADRBK2 (Uniprot code: P35626), GAD1 (Uniprot code: Q99259), GAD2 (Uniprot code: Q05329), HSPD1
(Uniprot code: P10809), SLC30A8 (Uniprot code: Q8IWU4). and SLC33A1 (Uniprot code: O00400). were retrieved from AAgAtlas.
The Ellipro tool predicted several possible cross-reactive epitopes between autoantigens and bacterial antigens (Table 1; Figure
1-6).
Conclusion: Bacterial antigens that mimic the autoantigens involved in Cystic Fibrosis have been identified, suggesting that
there is a relationship between infections and the development of this disease or the development of bacterial resistance, which
could explain the etiology in some patients.
KEYWORDS
Molecular mimicry; Cross reaction; Cystic fibrosis; P. aeruginosa; S. aureus
INTRODUCTION
To date, it is known that this condition, cystic fibrosis, is mainly caused by a genetic mutation, through a gene located on chromosome 7, encoding a protein that regulates transmembrane conductance (CFTR), which functions as a chloride activated by transmembrane cAMP [1]. There are many different genetic mutations in the CFTR gene, approximately there are more than 2,000 mutations, but for a better understanding of the pathophysiology they can be classified into 5 large groups such as: Defective protein synthesis, Defective protein processing, Disordered regulation, Chloride conductance defective and accelerated rotation of the channel [1,2]. One of the main mutations found among white American patients with Cystic Fibrosis, and in two-thirds of all registered patients in the world, is the F508 delta mutation.
Class 1 dysfunction is the result of a nonsense, frameshift, or
splice site mutation, leading to premature termination of the mRNA
sequence.
Class 2 dysfunction results in abnormal post-translational
processing of the CFTR protein. This step-in protein processing is
essential for proper intracellular transit of the protein [3].
Class 3 dysfunction is characterized by decreased protein
activity in response to intracellular signaling.
Class 4 dysfunction is when the protein is properly produced
and localized to the cell surface. However, the rate of chloride ion
flux and the duration of channel activation after stimulation are
decreased from normal [4].
Class 5 dysfunction is the net decrease in the concentration of
CFTR channels in the cell membrane as a result of rapid degradation
by cellular processes [5,6].
The result of all mutations is decreased chloride secretion and, consequently, increased sodium reabsorption in the cell space. Increased sodium reabsorption leads to increased water reabsorption and manifests as thicker mucous secretions in epithelial linings and more viscous secretions from exocrine tissues [6-8]. The most commonly affected organs include the sinuses, lungs, pancreas, biliary and hepatic systems, intestines, and sweat glands [9]. As reported in the literature, the disease occurs as a cascade effect following infection and the subsequent inflammatory process. Mucus plugging in the bronchioles results in a clinical picture of obstructive pulmonary disease. As a result of the obstruction, an optimal environment for bacterial growth is created within the airways. Bronchiectasis and further production of thick purulent sputum occur [10-12]. Part of the inflammatory reaction includes the production of interleukin-8 from neutrophils from epithelial cells, which functions as a secretagogue, increasing mucous secretion, thus creating a positive feedback loop of mucous secretion with persistent inflammation, infection, and structural damage [13]. The most common bacteria that cause endobronchial infection are Staphylococcus aureus [70% in childhood], and Pseudomonas aeruginosa [63% in adults] [13]. The observed increase in multiresistant Pseudomonas strains, as well as other rarer bacteria such as Burkholderia, Achromobacter, Stenotrophomonas and atypical mycobacteria, currently poses a therapeutic and hygienic challenge [13,14]. For this reason, it is desired to carry out this work, in order to analyze the molecular mimicry between this disease and the most frequent pathogens, and at the same time predict the possible cause of autoimmunity in patients with Cystic Fibrosis who are infected with bacteria such as P. aeruginosa and S. aureus. Molecular mimicry suggests the existence of antigens that are similar or identical to microorganisms and this autoimmune mechanism allows the microorganism to evade the immune response created by the host, subsequently allowing recognition of autoantigens after an infection that can develop in the host. person through cross-reactivity [15,16]. B and T lymphocytes can recognize pathogens and, at the same time, autoantigens. This mimicry mechanism means that it does so in four different ways: Through identical amino acid sequences, homologous but not identical sequences, common or similar amino acid sequences of different molecules (proteins and carbohydrates), and structural similarities between the microorganism or environmental agent and his guest [17]. Various data support molecular mimicry as a trigger for B cell-mediated autoimmunity in some cases; for example, antibodies have been identified against infectious organisms that show cross-reactivity with auto molecules associated with specific autoimmune diseases. Interesting examples are the observed cross-reactivity between Streptococcus pyogenes M protein and myosin in rheumatic heart disease, the cross-reactivity between Campylobacter and aquaporin [17].
In this study we have made progress in determining the molecular mimicry between the autoantigens involved in Cystic Fibrosis and the bacterial antigens involved in the development of the autoimmune response. To do this, an in-silico approach involving the recovery of amino acid sequences was used. Candidates for molecular mimicry were selected to predict cross-reactive epitopes if they share at least 30% amino acid sequence identity [18]. Bioinformatics allows the prediction, study, comparison of sequences and manages to propose candidate antigens as possible environmental triggers. Based on the facts mentioned above in this study, an exploratory in-silico analysis of bacterial antigens causing the possible development of autoimmunity secondary to Cystic Fibrosis and the generation of bacterial resistance is carried out.
MATERIALS AND METHODS
Selection of Autoantigens
Amino acid sequences of autoantigens for cystic fibrosis were selected from the AAgAtlas autoantigen database (http://biokb. ncpsb.org/aagatlas/). The amino acid sequence of each autoantigen was used as input in PSI-BLAST (https://blast.ncbi.nlm.nih.gov/ Blast.cgi) to find similar antigens of pathogens associated with infection in cystic fibrosis. Those selected to perform PSI-BLAST were Pseudomonas aeruginosa and Staphylococcus aureus. Only the antigens with similarity greater than or equal to 30% were selected for further analysis [19].
Epitope Prediction
For epitope prediction, two approaches were used: first, all autoantigens analysed in this study were used as input to the Ellipro server (http://tools.iedb.org/ellipro/), setting all parameters as defaults. Second, the potential epitope mapped for any autoantigen used here in this study was checked against the Immune Epitope Database (IEDB) (https://www.iedb.org/) [20,21].
RESULTS
Selection of Antigens
The autoantigens ADRBK2 (Uniprot code: P35626), GAD1 (Uniprot code: Q99259), GAD2 (Uniprot code: Q05329), HSPD1 (Uniprot code: P10809), SLC30A8 (Uniprot code: Q8IWU4), and SLC33A1 (Uniprot code: O00400) are retrieved from AAgAtlas, were reported as autoantibody targets in Cystic Fibrosis pathology. PSI-BLAST and binary alignment analysis found multiple matches at 30% identity with pathogen antigens (Table 1).
Alignments for the Prediction of Cross-Reactive Epitotes
The Ellipro tool predicted several potential cross-reactive epitopes between the ADRBK2 autoantigen and the P. aeruginosa serine/threonine protein bacterial antigen, GAD1 autoantigen and the P. aeruginosa aspartate aminotransferase family protein bacterial antigen, GAD1 autoantigen and the P. aeruginosa pyridoxal-bacterial antigen. dependent decarboxylase antigen from S. aureus, autoantigen GAD2 and the bacterial antigen aspartate aminotransferase family protein from P. aeruginosa, autoantigen HSPD1 and the bacterial antigen chaperonin GroEL from P. aeruginosa, and autoantigen HSPD1 and the bacterial antigen chaperonin GroEL from S. aureus (Figure 1-6). Binary alignment between ADRBK2 and P. aeruginosa showing a possible prediction of cross-reactivity in the epitope located at sequence 320 and possibly at 360 (Figure 1). Binary alignment between GAD1 and P. aeruginosa showing a possible prediction of crossreaction in the epitope located in sequence 340-350 and in 370 (Figure 2). Binary alignment between GAD1 and S. aureus showing a possible prediction of cross-reactivity in the epitope located at sequence 370 (Figure 3). Binary alignment between GAD2 and P. aeruginosa showing a possible prediction of cross-reactivity in the epitope located in the sequence 340 and 360-370 (Figure 4). Binary alignment between HSPD1 and P. aeruginosa showing a possible prediction of cross-reactivity in the epitope located in the sequence 60, 80, 110-120, 190, 220-230, 300, 430 and 530 (Figure 5). Binary alignment between HSPD1 and P. aureus showing a possible prediction of cross-reactivity in the epitope located in the sequence 30, 60, 80, 110-120, 220, 270-280, 300, 420, 430, 570 (Figure 6). For other autoantigens involved in Cystic Fibrosis and analysed in this study, we found that they share identity in their amino acid sequences with pathogenic antigens, such as: ADRBK2/ Chain B, Zpa963, cAMP-dependent protein or serine/threonine protein of Pseudomonas aeruginosa (30.7 and 31.9% correspondingly), SLC30A8/cation diffusion facilitator family transporter or cation transporter from Pseudomonas aeruginosa (33% and 34% correspondingly), SLC30A8/cation transporter from Staphylococcus aureus (34.6%) and SLC33A1/MFS transporter, partial or RhtX/ FptX family siderophore transporter of Pseudomonas aeruginosa (40.2% and 43% correspondingly) (Table 1).
DISCUSSION
Humanity’s fight against infectious diseases is well known. The discovery of antibiotics led to optimism that infections can be controlled and prevented. Yet infections remain the leading cause of death in the developing world [8]. This is due to the appearance of new diseases, the reappearance of diseases once controlled and, more specifically, the appearance of resistance to antimicrobials. The emergence of antimicrobial resistance appears to be unavoidable for almost all new drugs and is recognized as a major problem in the treatment of microbial infections both in hospitals and in the community [10,22]. Since ancient times, children all over the world have been affected by cystic fibrosis leading to shorter life expectancy. Salty skin was a sign of an impending illness with no cause or cure. Until relatively modern times, cystic fibrosis was poorly understood. In 1949, Lowe et al. postulated that cystic fibrosis must be caused by a genetic defect in the autosomal recessive pattern of inheritance of the disease. High salt levels in the sweat of cystic fibrosis patients suggested an abnormality in electrolyte transport from the sweat gland [22,23]. Researchers now know that cystic fibrosis is an autosomal recessive disorder of exocrine gland function that most commonly affects people of northern European descent at a rate of 1 in 3,500. It is a chronic disease that often leads to chronic sinopulmonary infections and pancreatic insufficiency. The most common cause of death is endstage lung disease [14,24].
In this study we have identified 6 autoantigens belonging to cystic fibrosis that share molecular mimicry with 2 bacterial antigens that frequently colonize patients with Cystic Fibrosis, among which are ADRBK2, GAD1, GAD2, HSPD1, and others (Table 1). This is the first report that relates bacterial infections as triggers of autoimmunity and bacterial resistance in cystic fibrosis at the in-silico level. The methodology implemented in this work is appropriate to describe the molecular mimicry between Cystic Fibrosis antigens and bacterial antigens at the in-silico level. Different approaches have been implemented, such as comparisons between whole proteomes, an example of this was provided by Bhardwaj T et al, in which they characterized the molecular mimicry between C. botulinum and the human proteome and this allowed the identification of antigens, such as enabled homologue and ATP-dependent DNA helicase, the 88C-containing coiled-coil domain, and the hypothetical protein [25]. It is also necessary to point out that our study has some limitations that could explain the lack of cross-reactivity between the autoantigens and the pathogen proteins evaluated. The present work is given by an insilico analysis, so it must be validated at the in vitro level, to fulfil complete criteria to establish the pathogens mentioned here as triggers of cystic fibrosis or bacterial resistance. However, in-silico analyses have many advantages to prioritize research resources and serve in the initial evaluation of a hypothesis as a support to define if further analysis is warranted.
CONCLUSION
In conclusion, bacterial antigens that mimic the autoantigens involved in Cystic Fibrosis have been identified, suggesting that there is a relationship between infections and the development of this disease, which could explain the aetiology in some patients. This supports what the literature shows, that after suffering from certain types of infections, patients could also develop autoimmunity. These findings support the need for meaningful clinical care in patients with bacterial infection and could be a predictive way of diagnosing CF. This study is of great clinical relevance. Since there is a present of molecular mimicry, we can be clear that the probability that there is some type of autoimmune disease associated with these pathogens is high, therefore we must be careful in patients who develop cystic fibrosis with the presence of these pathogens, in order to avoid an autoimmune disease and possible complications.
REFERENCES
- Lyamin AV, Ismatullin DD, Zhestkov AV, Kondratenko OV (2018) The laboratory diagnostic in patients with mucoviscidosis: a review. Klin Lab Diagn 63(5): 315-320.
- Poncin W, Lebecque P (2019) Lung clearance index in cystic fibrosis. Rev Mal Respir 36(3): 377-395.
- Bianconi I, D’Arcangelo S, Esposito A, Benedet M, Piffer E, et al. (2018) Persistence and microevolution of pseudomonas aeruginosa in the cystic fibrosis lung: a single-patient longitudinal genomic study. Front Microbiol 9: 3242.
- Awatade NT, Wong SL, Hewson CK, Fawcett LK, Kicic A, et al. (2018) Human primary epithelial cell models: promising tools in the era of cystic fibrosis personalized medicine. Front Pharmacol 9:1429.
- Pawlaczyk KT, Borysewicz LM, Śniatała R, Batura GH, Cofta S (2019) Dental and periodontal manifestations in patients with cystic fibrosis - A systematic review. J Cyst Fibros 18(6): 762-771.
- Radovanovic D, Santus P, Blasi F, Sotgiu G, D’Arcangelo F, et al. (2018) A comprehensive approach to lung function in bronchiectasis. Respir Med 145: 120-129.
- Fenker DE, McDaniel CT, Panmanee W, Panos RJ, Sorscher EJ, et al. (2018) A comparison between two pathophysiologically different yet microbiologically similar lung diseases: cystic fibrosis and chronic obstructive pulmonary disease. Int J Respir Pulm Med 5(2): 98.
- Rajapaksha IG, Angus PW, Herath CB (2019) Current therapies and novel approaches for biliary diseases. World J Gastrointest Pathophysiol 10(1): 1-10.
- (2017) Cystic fibrosis foundation. Drug development pipeline.
- Alton EW, Armstrong DK, Ashby D, Katie JB, Diana B, et al. (2015) Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med 3: 684-691.
- Davies JC, Cunningham S, Harris WT, Allen L, Warren ER, et al. (2016) Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med 4: 107-115.
- Quittner AL, Abbott J, Georgiopoulos AM, Lutz G, Beth S, et al. (2016) International committee on mental health in cystic fibrosis: cystic fibrosis foundation and European cystic fibrosis society consensus statements for screening and treating depression and anxiety. Thorax 71: 26-34.
- Rosenfeld M, VanDevanter DR, Ren CL, Eric PE, David JP, et al. (2015) Decline in lung function does not predict future decline in lung function in cystic fibrosis patients. Pediatr Pulmonol 50: 856-862.
- Griese M, Costa S, Linnemenn RW, Marcus AM, McKone EF, et al. (2020) Safety and efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 weeks or longer in people with cystic fibrosis and one or more F508del alleles: interim results of an open‐label phase 3 clinical trial. Am J Respir Crit Care Med 3(3): 381‐385.
- Filippi CM, von Herrath MG (2010) 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: viruses, autoimmunity and immunoregulation. Clin Exp Immunol 160(1): 113- 119.
- Mccarron A, Donnelley M, Parsons D (2018) Airway disease phenotypes in animal models of cystic fibrosis. Respir Res 19(1): 54.
- Dreano E, Bacchetta M, Simonin J, Louise G, Claire U, et al. (2019) Characterization of two rat models of cystic fibrosis‐KO and F508del CFTR‐Generated by Crispr‐Cas9. Animal Model Exp Med 2(4): 297‐311.
- Mccatton A, Parsons D, Donnelley M (2021) Animal and cell culture models for cystic fibrosis: which model is right for your application? Am J Pathol 191(2): 228‐242.
- Wang D, Yang L, Zhang P, LaBaer J, Hermjakob H, et al. (2017) AAgAtlas 1.0: a human autoantigen database. Nucleic Acids Res 45(D1): D769-D776.
- Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, et al. (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 9: 514.
- Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, et al. (2015) The immune epitope database (IEDB) 3.0. Nucleic Acids Res 43(Database issue): D405-D412.
- Li H, Zhu T, Chen M, Yang Y, Zhang H, et al. (2020) Cystic fibrosis in children with pseudo-Bartter syndrome: two cases report. J Wenzhou Med Univ 50(12): 1015‐1017.
- Qiu L, Yang F, He Y, Yuan H, Zhou J (2018) Clinical characterization and diagnosis of cystic fibrosis through exome sequencing in Chinese infants with Bartter‐syndrome‐like hypokalemia alkalosis. Front Med 12(5): 550‐558.
- Hauser AR (2015) Cell envelope. Antibiotic Basic for Clinicians (2nd Edn), Wolters Kluwer, New Delhi. India, Pp. 3-5.
- Fresqueta MSJR, Jowitta TA, McKenzie EA, Robertsc L, Lennon R, et al. (2020) Autoantigens PLA2R and THSD7A in membranous nephropathy share a common epitope motif in the N-terminal domain. Journal of Autoimmunity 106(102308).
Article Type
Research Article
Publication history
Received Date: August 25, 2022
Published: September 30, 2022
Address for correspondence
Janer Antonio Mora Lopez, Specialist in Critical Medicine and Intensive Care, Fundación Universitaria Ciencias de la Salud, Bogotá, Colombia, https://orcid.org/0000-0002-1039-2584
Copyright
©2022 Open Access Journal of Biomedical Science, All rights reserved. No part of this content may be reproduced or transmitted in any form or by any means as per the standard guidelines of fair use. Open Access Journal of Biomedical Science is licensed under a Creative Commons Attribution 4.0 International License
How to cite this article
Janer AML, Jhon FBC, Urbano DBR, Mario AGV, Fernan ATH, et al. Molecular Mimetism Between Cystic Fibrosis and P. Aeruginosa and S. Aureus, Possible Cause of Autoimmunity? In-Silico Analysis. 2022- 4(5) OAJBS.ID.000493.
Figure 1: Alignment of ADRBK2 autoantigen and P. aeruginosa bacterial serine/threonine protein antigen.
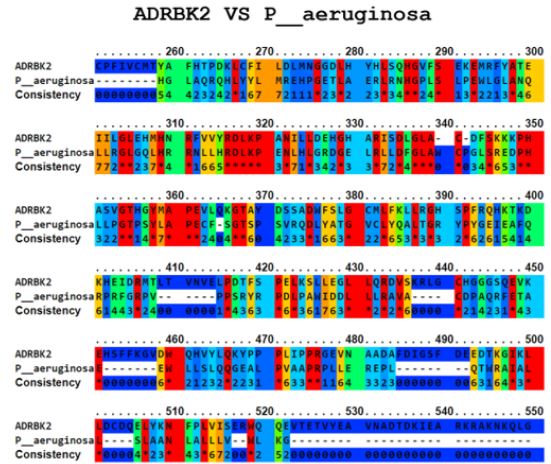
Figure 2: Alignment of the autoantigen GAD1 and the bacterial antigen aspartate aminotransferase family protein of P. aeruginosa.
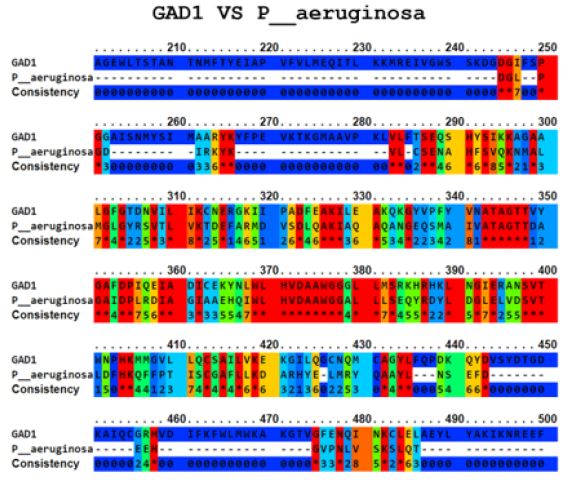
Figure 3: Alignment of the autoantigen GAD1 and the bacterial antigen pyridoxal-dependent decarboxylase of S. aureus.
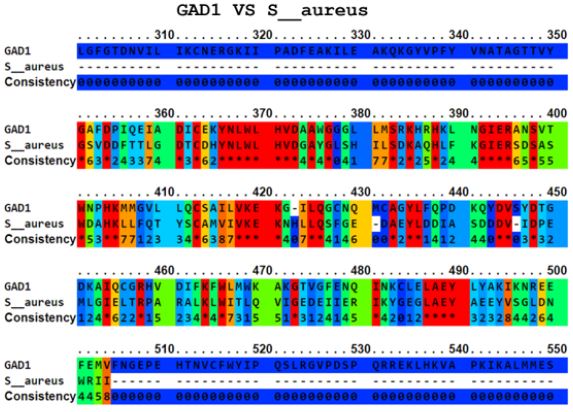
Figure 4: Alignment of the autoantigen GAD2 and the bacterial antigen aspartate aminotransferase family protein of P. Aeruginosa.
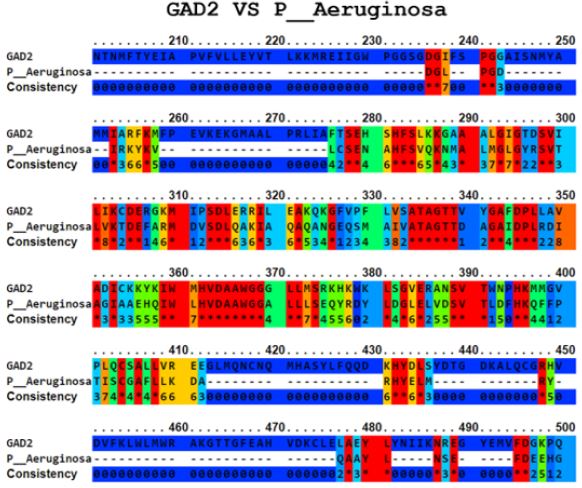
Figure 5: Alignment of the HSPD1 autoantigen and the bacterial antigen chaperonin GroEL from P. aeruginosa.

Figure 6: Alignment of the HSPD1 autoantigen and the bacterial antigen chaperonin GroEL from S. aureus.
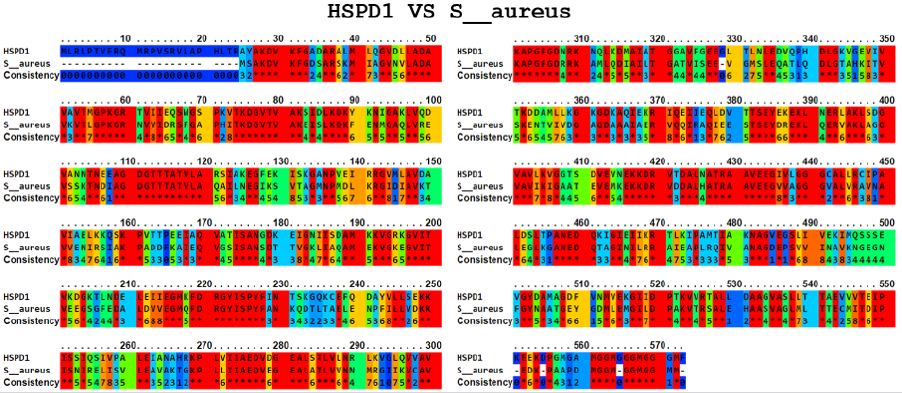
Table 1: List of cystic fibrosis autoantigens and bacterial antigens with molecular mimicry.