Core Proteomics Functional Analysis of Hypothetical Proteins of the Emerging Tilapia Lake Virus
ABSTRACT
TiLV is a single-stranded RNA virus with a negative sense that belongs to the Amnoonviridae family, genus Tilapinevirus, and species Tilapia tilapinevirus. The virus attacks the fish species’ essential organs, such as the eyes, brain, and liver. Syncytial cell development and widespread hepatocellular necrosis with pyknotic and karyolytic nuclei in the liver cells of infected fish are prominent pathological findings of this disease. The sickness is highly contagious, and it spreads both horizontally and vertically. In this study, we used the capabilities of multiple bioinformatics tools to assign probable functions for HPs based on protein family comparison, motifs, functional qualities of amino acids, and genomic context. The study excludes protein sequences containing less than 200 amino acid residues. Even though we were able to correctly estimate the function of 112 proteins. Enzymes, transporters, binding proteins, cell division, cell regulation, and other proteins were identified among the anticipated HPs. The findings of the study should help researchers better understand the molecular mechanisms underlying virus pathogenicity, as well as identify prospective targets for drug and vaccine development.
KEYWORDS
TiLV; HPs; Annotation; Characterization; Domain
INTRODUCTION
Millions of people throughout the world rely on fish and fisherybased activities for food, nutrition, money, and livelihood. The aquaculture business is becoming increasingly important to global food security, particularly in low-income, food-deficit countries, where it also helps to alleviate poverty. Given the restrictions of further expanding cattle pastures and the fact that the oceans are overfished, it has been suggested that aquaculture is the next and possibly final large-scale animal protein generating business. Indeed, aquaculture is the only animal food-producing industry that is increasing faster than the human population, and it is estimated that by 2030, 62% of the seafood we consume will have been reared on farms. Aquaculture will help food security if it receives long-term support. Aquaculture, on the other hand, has rapidly increased, intensified, and varied. When fish are moved from their natural home to the crowded and stressed environment of aquaculture, virulent wild-type viruses typically thrive [1,2]. In addition, Bacteria (55%), viruses (23%), parasites (19%), and fungi (3%) are the most common causes of infectious illnesses in aquaculture, according to the study. Tilapia lake virus (TiLV) is a newly discovered virus in Asia, Africa, Central America, and South America that causes enormous mortality in cultivated and feral tilapia (Oreochromis spp. and hybrids). Since 2014, there have been global reports of TiLV disease in tilapia fry, juveniles, and adults, leading to 10–90% mortality and huge economic losses. Besides, Tilapia tilapinevirus, often known as the tilapia lake virus, is the only species in the Tilapinevirus genus [3]. This orthomyxo-like virus has ten negativesense RNA segments encased within a membrane-bound nucleocapsid, despite not yet being classified to a viral family [4]. The viral particle is generally round in shape, with a diameter of 55 to 100 nanometers [5,6]. Because of its outer lipid membrane, TiLV is susceptible to organic solvents like ether and chloroform [7]. Although the amount of time TiLV can survive outside of its host has not been defined, experiments with tilapia have shown that it can be disseminated horizontally [7,8] and that it can survive in both freshwater and brackish water OIE 2018. Recent technological advancements in Next Generation Sequencing (NGS) generate more genomic data from a broader spectrum of viral and bacterial species. However, only a few bacterial and viral species have complete proteome information, but understanding disease etiology, virulence determinants, and their survival and spread require a thorough understanding of the microbial proteome Singh 2015. For example, about 30-40% of genes in the genomes of most viruses are labeled as unknown or speculative Hoskeri 2010. Hypothetical Proteins are proteins with uncertain functions or conserved hypothetical proteins that show only a limited relationship with functionally annotated proteins (HPs). HPs have translated parts of nucleic acid sequences based on sequence similarity, but functional and biochemical characterization must be evaluated for experimental existence Shahbaaz 2013. HPs also comprise low-identity proteins, as well as poorly characterized and ambiguous functional proteins Hoskeri 2010. HPs are divided into two groups: (i) Uncharacterized Protein Families (UPFs) and (ii) Domains of Unknown Function (DUFs).
UPFs are proteins that have been structurally identified but not functionally defined or connected to a known functional gene. Those whose experimental presence of a protein is known but is unrelated to any known functional or structural domain are referred to as DUFs. Annotating HPs has several benefits, including the ability to derive new structure-function relationships, unique protein structures, and the ability to decode additional pathways for a better knowledge of infections. HPs with functional annotations could be used as biomarkers for disease screening and diagnosis.
They can also be utilized to design and discover new drugs as a pharmacological target Singh 2015. Phylogenetic profiling, mass spectrometry identification, SAGE analysis, lethal analysis, and other methods can be used to predict the function of a protein [9]. Various computational methods have been created to facilitate the functional annotation process in order to reduce time and investment costs [10].
We employed the power of computational techniques to annotate the putative functions for the HPs in the Tilapia Lake virus in the current investigation. As a result, we used a databases and different bioinformatics tools to analyze the hypothetical proteins and reveal their functions. Computational techniques were widely used in the successful annotation of proteomes Shahbaaz 2013; Singh 2015. To completely comprehend the pathogenicity and genome flexibility of the Tilapia Lake Virus, it is necessary to decipher the role of entire gene coding areas in the genome.
METHODS AND MATERIALS
Sequence Retrieval
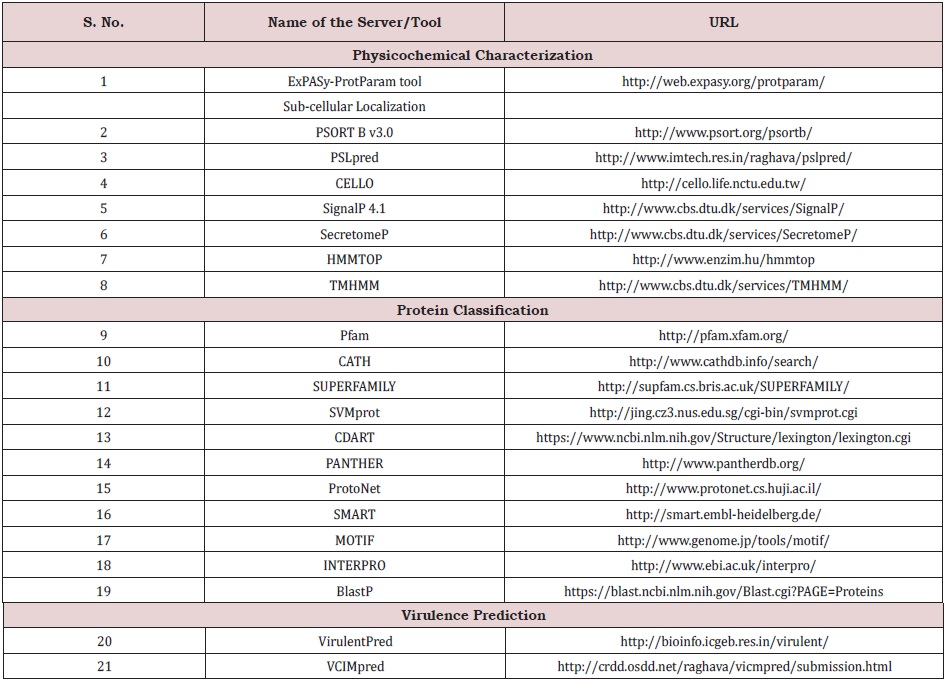
By examining the NCBI (http://www.ncbi.nlm.nih.gov/) database, the full protein sequences of Tilapia Lake Virus were discovered. The TiLV genome has 336 protein-coding genes, 112 of which have been designated as HPs. Using multiple functional annotation servers, all of the HP’s sequences were retrieved for biological functional assignment. Proteins with more than 200 amino acid residues were chosen from this study to reduce the number of misinterpretations in the functional annotation pathway. Table 1 lists several tools utilized in the functional predictions TiLV of HPs.
Physicochemical Characterization
ExPASy proteomics methods were used to characterize the HPs in terms of their physicochemical properties Gasteiger 2005. For the 112 HPs in Tilapia Lake Virus, parameters such as molecular weight (Mw), theoretical isoelectric point (pI), grand average of hydropathicity (GRAVY), aliphatic, and instability index were calculated.
Protein Localization
Various methods, including PSORTb, were used to estimate where the HPs would be found [11], PSLpred [12], and CELLO Yu 2004. The SignalP service was used to anticipate the signal peptide Secretome Petersen 2011. HPs were found in the nonclassical secretory route using this method. TMHMM was used to predict transmembrane information about the HPs [13] and HMMTOP servers [14].
Domain Identification and Functional Annotation
The HPs’ functions were predicted utilizing web-based techniques. NCBI-Blast [15], SMART [16], EBI-Interproscan [17] and Motif [18]. The functional domains/motifs found in the HPs were identified using these methods). Pfam was used to identify HP categories at the family level [19], SCOP Superfamily [20], and PANTHE [21] families. The CATH database was used to predict the structural level classification of protein superfamilies [22]. SVMProt was used to forecast the HPs’ domain architecture Cai 2003, CDART [23], and ProtoNet tools [24]. Virulentpred and VCIM-pred were used to forecast the HPs’ virulence activity.
RESULTS AND DISCUSSION
Sequence Retrieval and Physicochemical Characterization
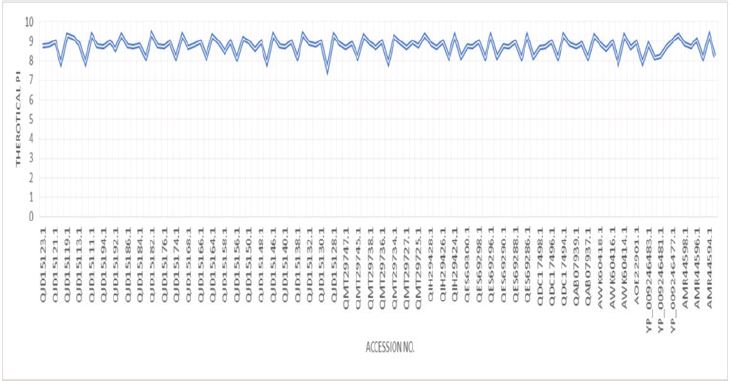
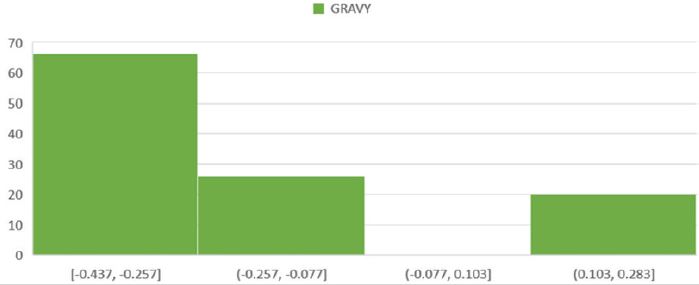
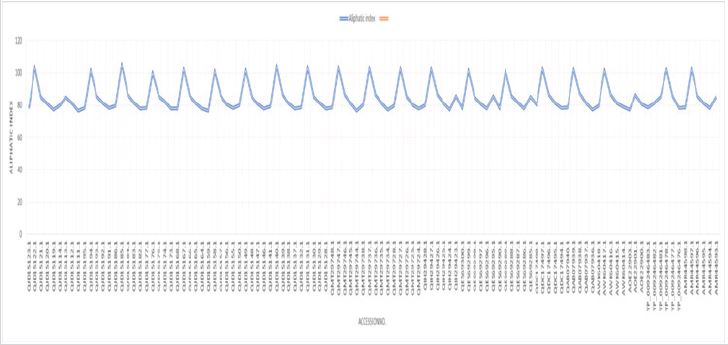
For physicochemical characterization and functional annotation, 112 HPs with more than 200 amino acid residues were chosen. The 112 HPs expected physicochemical attributes were summarized in Supplementary (Table 1) based on amino acid sequencing. In the functional characterization of proteins, molecular weight is an important parameter. The proteins QJD15123.1 and QES69285.1, with molecular weights (Mws) of 36223.65 and 57150.54 Da, respectively, have the lowest and highest Mws. The pH at which proteins have no net charge and mobility based on charge is zero is known as the isoelectric point (pI). Predicting pI is crucial in the construction of purification buffer systems and isoelectric focusing HPs had anticipated pI values ranging from 7.55 to 9.34. Seven proteins were found to be mildly basic out of 112 HPs, with projected pI values ranging from 7.55 to 7.91. Similarly, 105 proteins with pI values of 8.15 to 9.34 are classified as basic, and none have a neutral charge. The concentration of Cystine, Tryptophan, and Tyrosine amino acid residues in the protein sequences were used to calculate the HP’s extinguishment coefficient in water at 280nm. Extinction coefficient values are higher when these amino acid residues are more abundant in the protein. Extinction coefficient values were found to be very low in small proteins with 200 or more amino acid residues and a small number of Cystine, Tryptophan, and Tyrosine residues. In the drug development process, computing the extinction coefficient values aids in the quantitative investigation of protein-protein and protein-ligand interactions. The HPs instability index ranged from 38.93 to 59.98, with a low of 38.93 and a maximum of 59.98. The stability index value depicts the stability of the protein in a test tube environment. Only two proteins with an instability index less than 40 are categorized as stable, while 110 HPs with an instability index greater than 40 are classified as unstable. The aliphatic index is related to the number of aliphatic amino acid residues in the protein. Proteins with a higher aliphatic index, particularly globular proteins, have proved to be more thermally stable. The 112 HPs computed aliphatic index ranges from 76.43 to 104.84. The aliphatic amino acids (A, V, I, and L) play a major role in thermophilic protein stability, resulting in greater aliphatic index values to resist a wide range of temperatures. The GRAVY value represents the protein-water interactions and demonstrates the protein’s hydrophilic nature. The GRAVY values are calculated using the average sum of the amino acids hydrophilic and hydrophobic side chains. The lowest GRAVY value among the HPs is –0.437, while the highest is 0.274; (Figure 1-3); [25-37].
Subcellular Localization
The subcellular localization of proteins has been used to describe them as therapeutic and vaccination targets. Proteins found in the cytoplasmic matrix might be used as medication targets, while proteins found in the inner and outer membranes could be used as vaccination targets. The understanding of localization is a crucial step in determining the function of proteins. HPs were anticipated to be present in any of the cell’s locations (cytoplasm, periplasm, extracellular, inner membrane, and outer membrane) based on the knowledge of trained data sets. Based on the concurrence hits, we predicted the localization of 112 HPs out of 336 proteins for the study. 65.17% (73) of the anticipated 112 HPs were discovered to be present in the cytoplasm. HPs are found in 16.07 % of the inner membrane (18). In periplasm, 18.75 % (21) of HPs are expected to be present. The role of signal peptides in determining protein transport to the target site is crucial. As a result, knowing the transport system of certain proteins as well as the cleavage sites is critical for signal peptide prediction. Aside from a few cytoplasmic proteins, all other proteins contain signal peptides that aid in the transport of proteins across the membrane to their intended cellular site or organelles. Out of 112 HPs, we projected the presence of signal peptide sequences in 5 of them. Similarly, secretomes are proteins released by cells outside of the cell, and these proteins are vital for cellcell communication, cell proliferation, and disease. Membrane proteins are important in a variety of functions like transport, signaling, and energy transduction; as a result, membrane proteins account for half of the drug research targets. The ability to predict membrane proteins is critical for a better understanding of drug targets and the development of effective therapeutic compounds.
Functional Annotation
The function of HPs must be annotated to gain a better understanding of the entire biological system and design effective medications. We looked at 112 TiLV HPs to see what functions they might have. HPs were also examined for the existence of functional domains and signature motifs that were related to the biological function. We have successfully annotated the function of 112 HPs with high confidence. Supplementary (Table 1) shows a list of 112 proteins along with their associated functions. Only 33.33% of HPs functions were predicted based on the analysis, and the remainder HPs did not display concurrency findings, implying that a proper experimental technique must be coupled for improved functional assignment. Annotated proteins were majorly classified into the following six categories such as enzymes, binding proteins, cell division proteins, cell regulatory proteins, transporters, and the remaining proteins involved in different biological processes. However, we only found one binding protein, and the rest are proteins that were not predicted.
Binding Protein
Under binding proteins, we discovered 5 HPs. Influenza RNAdependent RNA polymerase subunit PB1 domain-like protein (QIH29423.1; QES69295.1; QES69285.1; YP 009246481.1; AMR44593.1), is made up of three subunits: P1, P2, and P3. In the interaction with the PB2 and PA subunits, the influenza virus PB1 protein has two distinct domains. The polymerase activity is controlled by PB1, which is at the center of the complex. Influenza RNA-dependent RNA polymerase subunit PB1 domain-like protein is well understood to have a vital part in a variety of processes. Transcriptional and replication of genomic RNA are both controlled by the RNA polymerase of influenza viruses.
RNA polymerase (transcription activity) was derived from insect cells carried with recombinant baculoviruses carrying cDNA for three P proteins. Various combinations of recombinant baculoviruses or nuclear extracts from insect cells infected with each of them or their transcription and replication abilities were investigated. Model template-directed RNA synthesis (an indicator of RNA replication) was facilitated by nuclear extracts of cells expressing all three P proteins (without primers), indicating that all three P proteins are necessary for RNA replication.
In all nuclear extracts including that containing only the PB1 subunit (an indication of transcription), template-directed dinucleotideprimed RNA synthesis was facilitated by the PB1 subunit. Researchers have discovered that PB1, and not PB2, or PA, is a catalytic subunit of influenza virus RNA polymerase and can catalyze RNA synthesise by itself.
Miscellaneous Protein
We grouped 107 hypothetical proteins that are involved in various biological processes into one category called other proteins added in the supplementary file. These proteins were unidentified and need further lab-based analysis to understand their characterization.
Virulence Proteins
To colonize the bacteria or virus, cause disease, and overcome the host’s defenses, virulence factors are required. As a result, knowing the molecular underpinnings of microbial virulence is critical for a virus’s pathogenesis. To predict the protein virulence factor among the 112 HPs, we employed VICMpred and VirulentPred, an SVM-based technique. Supplementary (Table 1) shows the projected findings. We discovered that 53 HPs are virulent factors, while 59 HPs are not. All of these proteins have the potential to be utilized as therapeutic targets in the future.
CONCLUSION
In general, roughly 30–50% of the sequenced genome is referred to as hypothetical proteins, and one of the main issues in modern biology is the rapid accumulation of genome data containing HPs. HPs obstruct the discovery of new therapeutic targets that can act directly on pathogens to combat pathogenicity. In the case of TiLV pathogens, these HPs are critical in identifying prospective therapeutic targets as well as improving our knowledge of their virulence and pathogenicity. Using multiple bioinformatics tools, we were able to characterize the roles of the 112 HPs from TiLV with a high level of confidence in this study. Various enzymes, transporters, cell division, and binding proteins have been identified as important in TiLV development, survival, virulence, and pathogenesis. Differentiating important therapeutic targets from vaccination targets is easier with protein characterization based on physiochemical characteristics and subcellular localization. Furthermore, we discovered 53 virulence proteins that are thought to be important in the pathogenesis of the organisms. As a result, future research on the projected HPs as novel therapeutic targets for drug and vaccine development may be aided by this study.
ACKNOWLEDGMENT
The first author is sincerely grateful to the ASEAN and Non-ASEAN scholarship authority at Chulalongkorn University, Thailand for giving financial support for pursuing master’s studies.
REFERENCES
- Hill BJ (2005) The need for effective disease control in international aquaculture. Dev Biol (Basel) 121: 3-12.
- Walker PJ, Mohan CV (2009) Viral disease emergence in shrimp aquaculture: origins, impact and the effectiveness of health management strategies. Reviews in Aquaculture 1(2): 125-154.
- King AMQ, Lefkowitz EJ, Mushegian AR, Adams, MJ Dutilh, et al. (2018) Changes to taxonomy and the International Code of Virus Classification and nomenclature ratified by the international committee on taxonomy of viruses (2018). Arch Virol 163(9): 2601-2631.
- Bacharach E, Mishra N, Briese T, Zody MC, Kembou Tsofack JE, et al. (2016) Characterization of a novel orthomyxo-like virus causing mass die-hffs of tilapia. MBio 7(2): e00431-16.
- Del-Pozo J, Mishra N, Kabuusu R, Cheetham S, Eldar A, et al. (2017). Syncytial hepatitis of tilapia (oreochromis niloticus L.) is associated with orthomyxovirus-like virions in hepatocytes. Veterinary Pathology 54(1): 164-170.
- Surachetpong W, Janetanakit T, Nonthabenjawan N, Tattiyapong P, Sirikanchana K, et al. (2017) Outbreaks of tilapia lake virus infection thailand, 2015-2016. Emerging Infectious Diseases 23(6): 1031-1033.
- Eyngor M, Zamostiano R, Kembou Tsofack JE, Berkowitz A, Bercovier H et al. (2014) Identification of a novel RNA virus lethal to tilapia. Journal of Clinical Microbiology 52(12): 4137-4146.
- Waiyamitra P, Piewbang C, Techangamsuwan S, Liew WC, Surachetpong W (2021). Infection of tilapia tilapinevirus in mozambique tilapia (oreochromis mossambicus. A Globally Vulnerable Fish Species Viruses 13(6).
- Gasperskaja E, Kučinskas V (2017) The most common technologies and tools for functional genome analysis. Acta Medica Lituanica 24(1): 1-11.
- Hawkins, Kihara, D. (2007). Function prediction of uncharacterized proteins. Journal of Bioinformatics and Computational Biology 5(1): 1-30.
- Bhasin M, Garg A, Raghava GPS (2005) PSLpred: prediction of subcellular localization of bacterial proteins. Bioinformatics 21(10): 2522-2524.
- Yu CS, Lin CJ, Hwang JK (2004) Predicting subcellular localization of proteins for gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sc: A Publication of the Protein Society 13(5): 1402-1406.
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of Molecular Biology 305(3): 567-580.
- Tusnády GE, Simon I (1998) Principles governing amino acid composition of integral membrane proteins: application to topology prediction. Journal of Molecular Biology 283(2): 489-506.
- Altschul SF, Gish W, Miller, Myers EW, Lipman, et al. (1990) Basic local alignment search tool. Journal of Molecular Biology 215(3): 403-410.
- Letunic I, Doerks T, Bork P (2012) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Research 40(D1): D302-D305.
- Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, et al. (2012). InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Research 40(D1) D306-D312.
- Kanehisa M (1997) Linking databases and organisms: Genome Net resources in Japan. Trends in Biochemical Sciences 22(11): 442-444.
- Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, et al. (2008). The Pfam protein family’s database. Nucleic Acids Research 36(1): D281-D288.
- Gough J, Karplus K, Hughey R, Chothia C (2001) Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. Journal of Molecular Biology 313(4): 903–919.
- Thomas PD, Kejariwal A, Guo N, Campbell MJ, Muruganujan A, et al. (2006) Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Research 34(2): W645-W650.
- Orengo CA, Michie AD, Jones S, Jones DT, Swindells, et al. (1997) CATH – a hierarchic classification of protein domain structures. Structure 5(8): 1093-1109.
- Geer LY, Domrachev M, Lipman DJ, Bryant SH (2002) CDART: protein homology by domain architecture. Genome Research, 12(10): 1619- 1623.
- Rappoport N, Karsenty S, Stern A, Linial N, Linial M (2012) ProtoNet 6.0: organizing 10 million protein sequences in a compact hierarchical family tree. Nucleic Acids Research 40(D1) D313-320.
- Cai CZ, Han L Y, Ji ZL, Chen X, Chen YZ, et al. (2003) SVM-Prot: Webbased support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Research 31(13): 3692–3697.
- Thilsted SH, Thorne-Lyman A, Webb P, Bogard JR, Subasinghe R, et al. (2016). Sustaining healthy diets: The role of capture fisheries and aquaculture for improving nutrition in the post-2015 era. Food Policy 61: 126–131.
- Bendtsen JD, Kiemer L, Fausbøll A, Brunak S (2005) Non-classical protein secretion in bacteria. BMC Microbiology 5(1): 58.
- Beveridge MCM, Thilsted SH, Phillips, MJ Metian, Hall SJ, et al. (2013) Meeting the food and nutrition needs of the poor: the role of fish and the opportunities and challenges emerging from the rise of aquaculture. Journal of Fish Biology 83(4): 1067–1084.
- Duarte CM, Marbá N, Holmer M (2007) Ecology. Rapid domestication of marine species. Science 316(5823): 382-383.
- Ferguson HW, Kabuusu R, Beltran S, Reyes E, Lince J A, et al. (2014) Syncytial hepatitis of farmed tilapia, oreochromis niloticus. a case report. Journal of Fish Diseases 37(6): 583-589.
- Kawarazuka N, Béné C (2011) The potential role of small fish species in improving micronutrient deficiencies in developing countries: building evidence. Public Health Nutrition 14(11): 1927–1938.
- Kibenge FS (2019) Emerging viruses in aquaculture. Current Opinion in Virology 34: 97-103.
- Kibenge FSB, Godoy MG, Fast M, Workenhe S, Kibenge MJT (2012) Countermeasures against viral diseases of farmed fish. Antiviral Research 95(3): 257–281.
- Roos N, Wahab MA, Chamnan C, Thilsted SH (2007) The role of fish in food-based strategies to combat vitamin A and mineral deficiencies in developing countries. The Journal of Nutrition 137(4): 1106-1109.
- Yada T & NakanishT (2002) Interaction between endocrine and immune systems in fish. International Review of Cytology 220: 35-92.
- Kembou Tsofack JE, Zamostiano R, Watted S, Berkowitz A, Rosenbluth E, et al. (2017) Detection of tilapia lake virus in clinical samples by culturing and nested reverse transcription -PCR. Journal of Clinical Microbiology 55(3): 759-767.
Article Type
Research Article
Publication history
Received Date: March 16, 2022
Published: May 04, 2022
Address for correspondence
Sk Injamamul Islam, Department of Fisheries and Marine Bioscience, Jashore University of Science and Technology, Bangladesh
Copyright
©2022 Open Access Journal of Biomedical Science, All rights reserved. No part of this content may be reproduced or transmitted in any form or by any means as per the standard guidelines of fair use. Open Access Journal of Biomedical Science is licensed under a Creative Commons Attribution 4.0 International License
How to cite this article
Moslema JM, Sk Injamamul IS. Core Proteomics Functional Analysis of Hypothetical Proteins of the Emerging Tilapia Lake Virus. 2022- 4(3) OAJBS.ID.000446.