Cerebellar Ataxia and Mesenteric Panniculitis – Possible Igg4-Related Disease, A Novel Presentation
ABSTRACT
IgG4-related disease is a multi-system inflammatory disease that may affect most systems. Intracranial parenchymal disease is rare, reported only in case reports. Meeting diagnostic criteria typically requires biopsy, creating diagnostic and treatment uncertainty if biopsy not possible. We present a case of likely IgG4-related disease causing both mesenteric panniculitis and acute cerebellitis, a presentation not known to be previously documented.
CASE REPORT
A 46-year-old man was transferred from a peripheral hospital for stroke workup, after presenting with sudden onset unsteadiness, left sided weakness and paraesthesia, nausea, vomiting and headache. He reported no facial droop, slurred speech, vertigo, or hearing loss. Other notable recent symptoms included coryzal symptoms several weeks ago attributed to allergic rhinitis and recent gardening, and a six-month deterioration in vision now requiring permanent bifocals, previously needed only for reading.
Relevant medical history included hypertension controlled with ramipril and lercanidipine, episodic chest pain with a negative exercise stress test, and obesity. Family history was positive for type two diabetes mellitus. The patient worked as a mechanical fitter with a history of occasional binge-pattern alcohol consumption, no recreational drug use, and ex-recreational cigarette smoking.
On examination, the patient was hypertensive at 180/120mmHg and afebrile. He was in sinus rhythm, with unremarkable ECG. On initial examination, he had bilateral infero-temporal quadrantopia, mildly reduced power on the left upper and lower limb with mild loss of superficial sensation on the lower left limb, and pronator drift on the left. Left sided plantar was upgoing, otherwise reflexes were appropriate. Cranial nerves examined as intact, with no change to optic disc on fundoscopy. He had profound unsteadiness of gait, which was wide-based and veering to the left side, with inability to tandem walk. There was past pointing on the left, though coordination was intact bilaterally on heelshin testing, no dysdiadochokinesia and Romberg’s negative.
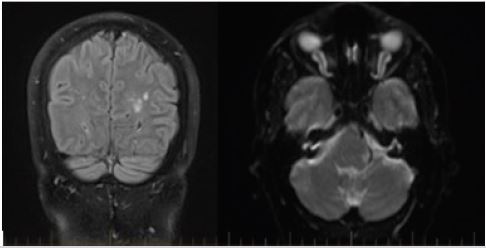
The patient was admitted to the medical ward for investigation of their neurological symptoms, and exclusion of acute cerebrovascular accident. Acute Disseminating Encephalomyelitis (ADEM), vasculitis and acute cerebellitis were primary differentials. A CT-stroke protocol noted no acute intracranial features, although profoundly patulous optic nerve sheath complexes seen bilaterally. Ongoing neurological symptoms prompted an MRI brain which identified extensive subcortical and periventricular hyperintense changes, seen best in Figure 1 the optic nerve sheath complexes were further assessed with a dedicated optic nerve MRI, noted to be swollen and fluid-filled.
Initial blood tests including inflammatory markers, thrombophilia screen and vasculitis screen were unremarkable. A carotid Doppler ultrasound and echocardiogram were also unremarkable. Focal neurological symptoms resolved within 48 hours of presentation, although ataxia persisted. Serum metanephrines, thyroid function and antibodies, and complement profile were unremarkable. ANA was 6, anticardiolipin <10, ANCA negative, RF <10, SSA, purkinje cell antibodies and aquaporin antibodies were not detected.
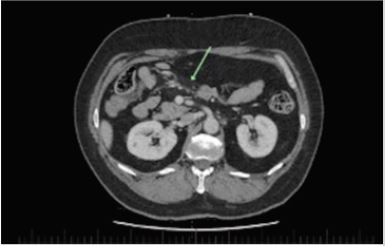
A lumbar puncture for CSF analysis demonstrated a protein level of 1.11, leukocytes 1x105 and 720 erythrocytes, with normal glucose and lymphocytes, oligoclonal bands not detected. PCR was negative for enterovirus, HSV 1 or 2, Neisseria meningitides and Varicella zoster. Paraproteins were not detected in urine protein electrophoresis. A PANCT identified mesenteric panniculitis (Figure 2) and an adrenal nodule. This nodule was assessed with a CT adrenal study and identified as a benign adrenal adenoma.
Multi-system inflammation in a middle-aged male prompted investigation of IgG4-RD. Serum IgG levels were noted to be elevated (17.3 g/L) and an IgG subclass panel identified an IgG4 of 1.67 (normal <1.35 g/L). A biopsy of the cerebellum, optic nerve sheath and mesentery to confirm an IgG4-RD diagnosis were not pursued. Acute cerebellitis was diagnosed on the clinical picture of dizziness, nausea, vomiting and ongoing gait ataxia with no loss of proprioception. No radiological inflammation or masses were seen on CT or MRI of the cerebellum.
A five-day course of daily 1g IV methylprednisolone was started nine days after presentation to the emergency department, given investigation findings with ongoing severe ataxia. On day two of treatment, the patient was able to walk unassisted. By day 3, he was able to tandem walk; cleared for independent mobilization on day 4; at baseline mobility by day 5. Subjective improvement in vision was also reported, described as “eyes are focusing more easily when I look into the distance”.
High dose steroids were initiated with the view to continuing immunosuppression after symptom remission. However, discussion with Neurological and Immunological specialties and our inability to confirm diagnosis on biopsy resulted in a steroid taper regimen following symptom remission. On day 12 of admission, the patient was discharged with Neurology and Ophthalmology outpatient follow up, on a prednisolone taper regimen to be managed by his primary care physician. Further follow-up has not been conducted at the time of paper submission.
DISCUSSION
IgG4-RD has been identified in almost every anatomical site, with a predilection for middle-aged patients, particularly males [1-4]. Intracranial IgG4-RD is uncommon, usually presenting as hypertrophic pachymeningitis, inflammatory pseudotumour, neuropathy and perineural disease, with few cases involving the parenchyma [2, 5-7].
We present a 46-year-old man with possible IgG4-RD affecting the cerebellum, optic nerve sheath and mesentery, the first case of IgG4-RD causing ataxia to our knowledge. A sparse number of cases have reported IgG4-RD involving the brain parenchyma without inflammatory pseudotumour or mass forming lesions [2,7]. One such case documented biopsy-proven parenchymal IgG4-RD presenting with focal neurology and dementia with white matter lesions hyperintense on T-2 MRI [3]. Radiological explanation for the patient’s immunosuppression-responsive dementia was not evident, suggesting inflammation of the parenchyma in IgG4-RD may not picked up on MRI [3].
Diagnosis of IgG4-related disease is a diagnostic challenge when biopsy is not obtained. The American College of Rheumatology (ACR) recently released three-step diagnostic criteria requiring 1) Classically affected organs, 2) Exclusion criteria composed of symptoms or pathology likely linked to other pathology and 3) A point system on histopathological, serum results and system involvement. Histopathology and immunostaining are critical criteria if suspected system involvement falls outside of the chest, pancreas/biliary tree, kidney and retroperitoneal arteries [8].
Where biopsy is unattainable, suggestive biochemistry or radiological features may be relied on to suggest diagnoses. Common differentials of IgG4-RD, such as malignancy, lymphoproliferative disorders, ANCA-vasculitis, sarcoidosis, Sjogren’s syndrome, and Castleman’s disease, may be ruled out with imaging and diseasespecific biochemistry, as performed for our patient [3-4,9]. CSF analysis may exclude important differentials such as malignancy and encephalitis. Classical findings in IgG4-RD are high protein with normal glucose and variable lymphocytosis.2 However, elevated CSF protein is also seen in GPA, RA, giant cell arteritis and sarcoidosis [5].
Elevated IgG4 levels are not diagnostic of IgG4-RD - high levels are seen in pathologies such as pancreatic cancer, autoimmune conditions, Castleman’s disease, ANCA-vasculitis and parasitic infections, and IgG4 may be normal in confirmed cases of IgG4- RD [1]. However, a meta-analysis found a sensitivity of 87.2% and specificity of 82.6% with serum IgG4 levels over 1.35 g/L, though the authors note significant heterogeneity [2]. A key feature of IgG4 disease is responsiveness to corticosteroid therapy, which may distinguish it from differentials such as Castleman’s disease [3,9- 10].
A treatment dilemma remains in cases where biopsy is unattainable, common differential diagnoses have been excluded, and biochemical and radiological tests suggest IgG4-RD as a possible diagnosis without another primary differential. Longterm management of IgG4-RD is immunosuppression with corticosteroids, or steroid-sparing agents such as mycophenolate and rituximab, all of which have significant side effects and possible morbidity [11]. Without a confirmed or probable diagnosis based on ACR criteria, treatment decisions after initial therapy are difficult. Follow-up by specialty physicians in the systems affected in individual patients is necessary to ensure appropriate follow-up.
IgG4-RD is considered a likely diagnosis due to our patient’s epidemiology, multi-organ inflammation, elevated IgG4 levels, rapid response to glucocorticoid therapy and exclusion of common IgG4-RD mimics [4]. Diagnosis was unable to be confirmed due to lack of biopsy. Despite lack of biopsy, our patient passed step one and step two of the ACR IgG4-RD diagnostic criteria but was unable to pass step three due to lack of biopsy, and unusual affected organs. Significant limitations in this diagnosis are noted, particularly failure to meet ACR criteria and atypical, if not unique, presentation of IgG4- RD. Despite these limitations, the authors suggest that in patients with undifferentiated neurological symptoms, IgG4-RD should be considered as a differential. Whilst isolated mesenteric panniculitis has been previously published as a manifestation of IgG4-RD with complete resolution with systemic steroid therapy [12-14], to best of our knowledge, mesenteric panniculitis and acute cerebellitis have not been previously described in combination.
CONCLUSION
In summary, IgG4-RD should be considered as a differential in the context of multi-system inflammation biochemically and/ or radiologically, including in the presence of undifferentiated neurological symptoms. American College of Rheumatology diagnostic criteria suggest IgG4-RD is very difficult to diagnose without biopsy. In cases where other differentials have been excluded and biopsy is not possible, consideration should be given to a steroid challenge to observe for response. All patients with possible IgG4-RD should have follow-up with appropriate subspecialties for their anatomical sites of inflammation.
REFERENCES
- Hao M, Liu M, Fan G, Yang X, Li J (2016) Diagnostic value of serum IgG4 for IgG4-related disease: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 95(21): e3785.
- Regev K, Nussbaum T, Cagnano E, Giladi N, Karni A (2014) Central nervous system manifestation of IgG4-related disease. JAMA Neurol 71(6): 767-770.
- Della-Torre E, Lanzillotta M, Doglioni C (2015) Immunology of IgG4- related disease. Clin Exp Immunol 181(2): 191-206.
- Al-Khalili OM, Erickson AR (2018) IgG-4 related disease: an introduction. Mo Med 115(3): 253-256.
- Goulam-Houssein S, Grenville JL, Mastrocostas K, Munoz DG, Lin A, Bharatha A, et al. (2019) IgG4-related intracranial disease. Neuroradiol J 32(1): 29-35.
- Fenderson J, Berenberg J, Tom L, Gress F (2015) IgG4-related disease: imitating a great imitator. Hawaii J Med Public Health 74: 22-26.
- Baptista B, Casian A, Gunawardena H, D’Cruz D, Rice CM (2017) Neurological manifestations of IgG4-related disease. Curr Treat Options Neurol 19(4): 14.
- Wallace ZS, Naden RP, Chari S, Choi HK, Della-Torre E, et al. (2020) The 2019 American College of Rheumatology/European League against rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol 72(1): 7-19.
- Otani K, Inoue D, Fujikura K, Komori T, Abe-Suzuki S, et al. (2018) Idiopathic multicentric Castleman’s disease: a clinicopathologic study in comparison with IgG4-related disease. Oncotarget 9(6): 6691-706.
- Mulholland GB, Jeffery CC, Satija P, Côté DW (2015) Immunoglobulin G4- related diseases in the head and neck: a systematic review. J Otolaryngol Head Neck Surg 44(1): 24.
- Brito-Zerón P, Kostov B, Bosch X, Acar-Denizli N, Ramos-Casals M, et al. (2016) Therapeutic approach to IgG4-related disease: a systematic review. Medicine (Baltimore) 95(26): e4002.
- Salvarani C, Valli R, Boiardi L, Pipitone N, Nicoli F, et al. (2011) IgG4- associated sclerosing mesenteritis. Clin Exp Rheumatol 29: S79-S80.
- Emory TS, Monihan JM, Carr NJ, Sobin LH (1997) Sclerosing mesenteritis, mesenteric panniculitis and mesenteric lipodystrophy: a single entity? Am J Surg Pathol 21(4): 392-398.
- Butt Z, Alam SH, Semeniuk O, Singh S, Chhabra GS, et al. (2018) A case of IgG4-related sclerosing mesenteritis. Cureus 10(2): e2147.
Article Type
Case Report
Publication history
Received date: July 08, 2021
Published date: July 26, 2021
Address for correspondence
Adel Ekladious, Associate professor of medicine, Faculty of health and medical sciences, University of Western Australia, 35/ Stirling Highway, Perth Western Australia
Copyright
©2021 Open Access Journal of Biomedical Science, All rights reserved. No part of this content may be reproduced or transmitted in any form or by any means as per the standard guidelines of fair use. Open Access Journal of Biomedical Science is licensed under a Creative Commons Attribution 4.0 International License
How to cite this article
Adel Ekladious, Emily Watter, James Allen. Cerebellar Ataxia and Mesenteric Panniculitis – Possible Igg4-Related Disease, A Novel Presentation. 2021- 3(4) OAJBS. ID.000307.