
Antibiotic Resistance of Pseudomonas aeruginosa Chronic Infection in Cystic Fibrosis Patients. A Single- Center Long-Term Follow-Up
ABSTRACT
Pseudomonas aeruginosa is the major bacterial pathogen associated with increased mortality and morbidity in cystic fibrosis patients. This study compared the antimicrobial susceptibilities of 56 Pseudomonas aeruginosa isolates collected from 28 Romanian pediatric patients over 15 years (2005-2020) and investigated the resistance to the usual antibiotics and the incidence of multidrug resistance Pseudomonas aeruginosa. The susceptibility rates to antimicrobials were as follows: Gentamicin 50%; Cefepime 53.6%; Ceftazidime 64.3%; Amikacin 67.9%; Ciprofloxacin 75%; Piperacillin 75%; Piperacillin-Tazobactam 82.1%; Meropenem 82.1% Levofloxacin 89.3%; Colistin 100%. Half of the patients gain antibiotic resistance during the study period. We noticed the most significant increase in antibiotic resistance for Ciprofloxacin, 14.3%, followed by Meropenem 10.8%, Ceftazidime 10.7%, and Piperacillin 10.7%. Multi-drug resistance Pseudomonas aeruginosa strains were identified with a frequency of 25% of 56 samples. 28.5% of patients with multi-drug resistance Pseudomonas aeruginosa strains were identified. Only one patient meets the criteria and can be classified as extensively drug-resistant Pseudomonas aeruginosa, sensitive only to Colistin. New therapeutic strategies for the treatment of Pseudomonas aeruginosa infections are an immediate necessity.
KEYWORDS
Cystic fibrosis; Pseudomonas aeruginosa infection; Inflammation; Multi-drug resistant Pseudomonas aeruginosa; Children; Colistin
INTRODUCTION
Cystic fibrosis (CF) is an autosomal recessive disease, occurring with approximately 30,000 patients in the USA and over 70,000 worldwide, which primarily affects the respiratory system and often leads to respiratory complications responsible for significant morbidity and mortality [1,2]. CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which encodes a protein [3]. This protein works as a channel across the membrane of cells, transports chloride ions into and out of cells and regulates sodium ions transport across cell membranes. Almost 2000 CFTR gene mutations have been identified, and the F508del mutation accounts for 50-70% of cases worldwide [4,5]. Dysfunction of this channel affects epithelial ion and water transport stability, causing mucus secretions to become thick and sticky. CFTR protein is localized in the apical membrane of secretory epithelial cells of exocrine glands [2,6]. CFTR dysfunction causes obstructions in the respiratory airways, inducing the accumulation of thick, viscous mucus in the respiratory tract, which prevents microbial clearance, generates a pro-inflammatory microenvironment, leading to chronic inflammation and infections [7]. It can affect the pancreas, the liver, the sweat glands leading to malabsorption, biliary cirrhosis, heat shock, and infertility [8].
Many factors, including geographic variation, ethnicity, type of genetic mutation, prolonged hospitalization, antibiotic exposures, and chronic infection, can affect the composition of the airway microbiome [9]. Staphylococcus aureus, methicillin-resistant Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa and Burkholderia cepacia have been recorded as the major respiratory pathogens in cystic fibrosis patients, but also nontraditional microorganisms like Achromobacter xylosoxidans, Stenotrophomonas maltophilia, non-tuberculosis Mycobacterium have been identified and associated with worsening lung function [10].
CF is characterized by chronic inflammation and polymicrobial infection. Pseudomonas aeruginosa infection can be described as a multi-stage process. Early infection with Pseudomonas aeruginosa may be easy to eradicate. The success rate was 90%, and this was explained due to initial acquisition with environmental strains, with predominantly non-mucoid and antibiotic susceptible isolates. Chronic infection is complicated to eradicate due to the development of a mucoid biofilm, and it is associated with increased mortality and morbidity [11-14]. Different eradication strategies have been proposed. Nebulized antibiotics such as Colistin or Tobramycin, alone or in combination with oral antibiotics such as Ciprofloxacin, were most frequently used. Several studies compare different strategies of inhaled, oral and intravenous antibiotics and report insufficient evidence to state which antibiotic strategy should be used to eradicate early Pseudomonas aeruginosa infection in cystic fibrosis [11,15]. Chronic infection occurs when there is a bacterial switch to a chronic mode of survival. Pseudomonas aeruginosa holds a high level of intrinsic resistance expressed by restricted outer membrane permeability, efflux systems that pump antibiotics out of the cell, and production of antibiotic-inactivating enzymes, along with the acquired resistance and biofilm formation, lead to multidrug-resistant strains and persistent infections [14]. Antibiotic resistance frequently develops in acute exacerbations leading to multiple hospitalizations or chronic infections, mainly when the frequent use of antibiotics facilitates the selection of multi-drug resistance Pseudomonas aeruginosa strains [16]. It has been described as resistant to various antibiotics, including aminoglycosides, quinolones, β-lactams, and carbapenems [17,18]. New therapies such as inhaled Levofloxacin, Amikacin Liposome have been shown to improve lung function and the quality of life by reducing the number of exacerbations [16]. New studies show good results regarding the use of Ceftolozan/ Tazobactam for the treatment of multi-drug resistance Pseudomonas aeruginosa [19,20].
This study aims to identify the resistance of Pseudomonas aeruginosa strains to the usual antibiotics, describe the frequency of multi-drug resistance strains isolated from the respiratory samples of CF patients, and evaluate antibiotic evolution resistance in the studied period.
MATERIALS AND METHODS
This study was conducted at the Pediatric Clinic of the M.S. Curie Children’s Emergency Clinical Hospital, Bucharest. It is a retrospective cohort observational study conducted over 15 years. We studied the medical history of 64 patients diagnosed with cystic fibrosis in the period 2005-2020.
Patients who tested positive for chloride sweat test and the diagnosis were confirmed by genetic testing were included. The sweat test is considered the “gold standard” for the diagnosis of CF. A sweat chloride concentration of 60 mmol/L or greater on two separate occasions is indicative of CF. Screening for CFTR mutations was performed initially by analyzing 38 mutations and the Allele5T-7T-9T polymorphism. If no CFTR mutation has been identified, then the whole-gene CFTR sequencing was performed. Respiratory specimens such as sputum or bronchoalveolar lavages were collected during an acute exacerbation or a periodic medical examination.
Patients who had at least two positive samples for Pseudomonas aeruginosa, followed by antibiotic susceptibility, were included. Routine culture for Pseudomonas aeruginosa was performed on blood agar and MacConkey agar. The media were incubated for 18-24 h at 37 °C, and Pseudomonas aeruginosa was identified by biochemical tests [21,22]. Antibiotic susceptibility testing was performed using the automatic method Vitek 2 in compliance with the European Committee on Antimicrobial Susceptibility Testing (EUCAST). The antibiotic susceptibility testing for aminoglycosides (Gentamicin and Amikacin), fluoroquinolones (Ciprofloxacin and Levofloxacin), beta-lactams (Ceftazidime, Cefepime, Meropenem, Piperacillin, Piperacillin-tazobactam), and polymyxin (Colistin) were performed by microdilution method [23-25]. Patients who had a positive sample for Pseudomonas aeruginosa and underwent early eradication therapy with subsequent negative samples were excluded. Also, all the patients without screening for CFTR mutations were excluded. Only 28 patients met the criteria mentioned above.
We performed statistical analysis and graphs using the Analyze IT 5.5 program (Microsoft Office Excel Add-on, Leeds, UK). Continuous variables had a non-gaussian distribution and were presented as the median and the interval between the 1st and 3rd quartiles in square brackets. The differences between the semiquantitative variables were assessed by the Mann- Whitney U test. Categorical variables were presented as numbers and percentages in round brackets. Differences in quantitative parameters were tested using nonparametric tests (Kruskal- Wallis). Qualitative data were compared with the chi-square test or Fisher exact test. We considered statistical significance at a p-value of 0.05 or lower. All patient data were anonymized before analysis.
Limitations of the study include the impossibility of studying the susceptibility of Pseudomonas aeruginosa infection to Tobramycin due to the lack of testing for the whole group and the impossibility to determine the phenotypic type (mucoid/nonmucoid) of Pseudomonas aeruginosa isolates (Figure 1).
RESULTS AND DISCUSSION
A total of 28 patients, median age 15.5 years [10; 20], 57.1 % males and 42.9% females, with a median age at diagnosis of 4 months [3; 26.2] and with BMI 16.65 kg/m2 [14.82; 19.65] were studied. The most common genetic mutation was heterozygous for F508del (57.1%), followed by homozygous for F508del (39.3%). Other mutations such as G542X (14.3%), W1282X (14.3%) and 349delTT (7.1%), G194R (7.1%), 1677delTA (7.1%), L1304X (7.1%), R347P (7.1%), R785X (7.1%), R1066H (7.1%), L88X/ G106PR (7.1%), 3272-26A-G (7.1%) were found. All patients had pancreatic insufficiency with median levels of stool elastase 17μg/g [13.19; 39.25]. Pancreatic insufficiency and CF liver disease are associated with low bone mineral density [26]. High levels of calprotectin [72.2; 1192ug/g] were noticed. Type 1 diabetes was found in 7 patients (25%). Cystic fibrosis-related diabetes (CFRD) is considered one of the most important comorbidities for CF patients. It is associated with an increased number of exacerbations and poor lung function; thereby, annual screening with an oral glucose tolerance test is recommended from the age of 10 [27,28].
The most frequent pathogens identified in the sputum and/ or bronchoalveolar lavages besides Pseudomonas aeruginosa were methicillin-resistant Staphylococcus aureus 57.1%, Staphylococcus aureus 39.3%, Klebsiella pneumoniae 28.6%, Escherichia coli 25%, Stenotrophomonas maltophilia 21.4%, Aspergillus fumigatus 17.9%, Burkholderia cepacia 14.3% and Mycobacterium tuberculosis 7.1%. Recurrent respiratory infections associated with obstructive lung disease are responsible for high morbidity and mortality rates due to respiratory insufficiency. It has been shown that Staphylococcus aureus and Haemophilus influenzae are more common in pediatric subjects, while Pseudomonas aeruginosa and Burkholderia cepacia are more common in older age. It has been noticed that Pseudomonas aeruginosa infection is associated with more severe lung disease [29], especially in female subjects [9,30]. Assessment of the lung function using spirometry showed median levels of FEV1 76.5% [63.2; 90.1], FVC 69.1% [60.5; 81.1], and an absolute FEV1/FVC ratio of 1.06 [0.98; 1.11], which indicates a restrictive ventilatory impairment [Table 1].
We studied 56 antibiotic susceptibility testing for Pseudomonas aeruginosa, at least two for each patient. The initial rates of susceptibility to antimicrobials at were as follows: aminoglycosides- Amikacin 67.9%, Gentamicin 57.1%; fluoroquinolones- Ciprofloxacin 89.3%, Levofloxacin 96.4%; beta-lactams-Ceftazidime 75%, Cefepime 53.6%, Piperacillin 85.7%, Piperacillin-Tazobactam 85.7%, Meropenem 92.9%; polymyxin- Colistin 96.4% [Table 2].
Multi-drug resistance Pseudomonas aeruginosa was defined as a non-susceptibility (intermediate plus resistance) to at least one agent in three or more antimicrobial categories: aminoglycosides, carbapenems, fluoroquinolones, penicillins+b-lactamase inhibitor, extended-spectrum cephalosporins, polymyxins, tetracyclines. Extensively drug-resistant (XDR) was defined as non-susceptibility to at least one agent in all categories, except two or fewer antimicrobial categories [31].
Half of the patients (50%) gain antibiotic resistance during the study period. The susceptibility rates to antimicrobials were lower than at the beginning of the study except for Amikacin and Cefepime, which had the same susceptibility rate. We noticed the most significant increase in antibiotic resistance for Ciprofloxacin, 14.3%, followed by Meropenem 10.8%, Ceftazidime 10.7%, Piperacillin 10.7%, Gentamicin 7%, Levofloxacin 7%, Piperacillin- Tazobactam 3.1%. An increase in antibiotic susceptibility has been identified only for Colistin [Figure 2].
Although we note a reduced antibiotic susceptibility of Pseudomonas aeruginosa during the study period, there are no results of statistical significance. The authors explain this partly due to correct targeted therapy and due to the small cohort studied. Lucca et al. [32] reported similar results, showing a significant decrease in antibiotic sensitivity for fluoroquinolones and an increased antibiotic sensitivity for Amikacin and Colistin. Fourteen sputa/bronchoalveolar lavage samples were positive for multidrug resistance Pseudomonas aeruginosa, given a frequency of 25%. Another study tested 61 samples of Pseudomonas aeruginosa isolated from 30 patients with cystic fibrosis and reported a frequency of 19.7% for multi-drug resistance Pseudomonas aeruginosa [25]. Several studies showed an increased incidence of multi-drug resistance organisms among respiratory isolates, reporting a rate of 73% methicillin-resistant Staphylococcus aureus and between 26% and 43% multi-drug resistance Pseudomonas aeruginosa [33]. 28.5% of patients with multi-drug resistance Pseudomonas aeruginosa strains were identified. Three patients maintained the initial antimicrobial resistance throughout the study, and the other three patients acquired new resistance to the following antibiotics: Ceftazidime, Ciprofloxacin, Gentamicin, Piperacillin-Tazobactam, Meropenem Amikacin. All these patients have pancreatic insufficiency with mean levels of stool elastase between 13.19-39.25 μg/g. Ideozu et al. [34] noticed that pancreatic insufficient CF subjects showed increased colonization with any form of Pseudomonas aeruginosa compared to the sufficient pancreatic group.
We mention two patients who presented multi-drug resistance Pseudomonas aeruginosa strains at the beginning of the study but not at its end. The authors explain this due to a correctly performed antibiotic treatment, as well as due to good compliance of the patient [35-37]. Moreau-Marquis et al. [38] reported that ΔF508 CFTR mutation is associated with increased Pseudomonas aeruginosa biofilm formation by increasing iron availability. In the airway of CF patients is an increased amount of iron, which is known to increase the growth of Pseudomonas aeruginosa in contrast to iron chelation which reduces biofilm production.
These patients were found to be ΔF508 heterozygotes, and those who maintained multi-drug resistance Pseudomonas aeruginosa strains during the study had the ΔF508 genetic mutation in the homozygous form. Several studies showed that patients with CF who are homozygous for the ΔF508 mutation usually have an increased risk of detecting Pseudomonas aeruginosa and thereby more severe disease. One patient meets the criteria and can be classified as extensively drug-resistant Pseudomonas aeruginosa, sensitive only to Colistin.
There was no increased antimicrobial resistance identified to Colistin throughout the study period, so we report a 100% susceptibility rate at the end of the study even though all patients received treatment with inhaled Colistin. This result is following the data in the literature. Ekkelenkamp et al. [39] tested 414 Pseudomonas aeruginosa isolates from respiratory samples of CF patients and reported only a percentage of 4% resistance to Colistin [40,41]. Mustafa et al. [42] studied 153 Pseudomonas aeruginosa isolates from 118 patients and reported a susceptibility rate based on EUCAST breakpoints of 92%. Jansen et al. [43] concluded that resistance to Colistin was rare despite its frequent use in the studied patients.
Infections with carbapenem-resistant Pseudomonas aeruginosa represent a significant public health problem due to a challenging treatment and possible widespread transmission. Worldwide increased carbapenem resistance ranges from 10% to 50% [42]. Our study emphasizes these results by reporting an increase in carbapenem resistance rate of 10.8%, surpassed only by Ciprofloxacin 14.3% and a rate of 25% multi-drug resistance Pseudomonas aeruginosa. In the 2019 surveillance report, the European Centre for Disease Prevention and Control reports the highest mean resistance percentage for fluoroquinolones, 18.9%, Piperacillin + Tazobactam 16.9%, and carbapenems 16.5%, and a mean percentage of 12.1% combined resistance [43].
Depending on the urgency of developing new antibiotic treatment, World Health Organization (WHO) listed carbapenemresistant Pseudomonas aeruginosa alongside carbapenem-resistant Enterobacteriaceae and carbapenem-resistant Acinetobacter baumannii as microorganisms with critical priority. In light of these findings, early eradication of Pseudomonas aeruginosa infection in cystic fibrosis is essential because antibiotic resistance frequently develops once the chronic infection is established. All the data provided by the studies are necessary to find a better strategy to manage these infections. Discoveries or alternative therapeutic strategies against this opportunistic agent are an immediate necessity. Although we note a reduced antibiotic susceptibility of Pseudomonas aeruginosa during the study period, there are no results of statistical significance. The authors explain this due to the small cohort studied.
CONCLUSION
Pseudomonas aeruginosa is the primary respiratory pathogen in cystic fibrosis patients, whose prevalence increases with age and whose eradication is increasingly difficult due to the appearance of multi-drug resistance and extensively drug-resistant strains. Half of the patients gain antibiotic resistance during the study period. The emergence of multi-drug resistance Pseudomonas aeruginosa was 25%. Ciprofloxacin and Meropenem had the most significant increase in resistance rate (14.3% versus 10.8%). One patient meets the criteria and can be classified as extensively drug-resistant Pseudomonas aeruginosa, sensitive only to Colistin; therefore, we report a 100% susceptibility rate. Considering that WHO listed Pseudomonas aeruginosa as one of the three bacteria with critical priority as a target of antibiotic therapy research, it is essential to emphasize that although it was introduced to Europe in the 1950s and used frequently for cystic fibrosis patients, Colistin remains a valid therapeutic option for treating multi-drug resistance and extensively drug-resistant Pseudomonas aeruginosa.
AUTHOR CONTRIBUTIONS
Conceptualization, Marcela Daniela Ionescu and Elena Camelia Berghea; Data curation, Maria Cristina Stefan and Maria Iulia Brustan; Formal analysis, Andrei Capitanescu; Investigation, Maria Iulia Brustan; Methodology, Mihaela Bălgrădean, Maria Cristina Stefan, Cristina Filip and Elena Camelia Berghea; Project administration, Marcela Daniela Ionescu, Mihaela Bălgrădean, Cristina Filip and Elena Camelia Berghea; Resources, Maria Iulia Brustan; Software, Andrei Capitanescu; Supervision, Mihaela Bălgrădean; Writing – original draft, Marcela Daniela Ionescu and Maria Cristina Stefan.
Institutional Review Board Statement
The research was approved by the Ethics Committee of “Marie Curie” Emergency Children’s Hospital
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
REFERENCES
- Regard L, Martin C, Chassagnon G, Burgel PR (2019) Acute and chronic non-pulmonary complications in adults with cystic fibrosis. Expert Rev Respir Med 13(1): 23-38.
- Ehsan Z, Clancy JP (2015) Management of Pseudomonas aeruginosa infection in cystic fibrosis patients using inhaled antibiotics with a focus on nebulized liposomal amikacin. Future microbiology 10(12): 1901- 1912.
- Fanen P, Wohlhuter-Haddad A, Hinzpeter A (2014) Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. The international journal of biochemistry & cell biology, 52: 94-102.
- (2020) U.S. National Library of Medicine. CFTR gene: MedlinePlus Genetics.
- Buchanan PJ (n.d.). Microbial infection in cystic fibrosis. British Society for Immunology.
- Vankeerberghen A, Cuppens H, Cassiman JJ (2002) The cystic fibrosis transmembrane conductance regulator: an intriguing protein with pleiotropic functions. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society 1(1): 13-29.
- Bhagirath AY, Li Y, Somayajula D, Dadashi M, Badr S, et al. (2016) Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC pulmonary medicine 16(1): 174.
- Elborn JS (2016) Cystic fibrosis. Lancet 388(10059): 2519-2531.
- Janahi IA, Rehman A (2017) The cystic fibrosis airway microbiome and pathogens. Progress in Understanding Cystic Fibrosis.
- Kiedrowski MR, Bomberger JM (2018) Viral-bacterial co-infections in the cystic fibrosis respiratory tract. Front Immunol 9: 3067.
- Langton Hewer SC, Smyth AR (2017) Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database Syst Rev 4(4): CD004197.
- Taccetti G, Campana S, Festini F, Mascherini M, Döring G (2005) Early eradication therapy against Pseudomonas aeruginosa in cystic fibrosis patients. Eur Respir J 26(3): 458-461.
- Kidd TJ, Ramsay KA, Vidmar S, Carlin JB, Bell SC, et al. (2015) Pseudomonas aeruginosa genotypes acquired by children with cystic fibrosis by age 5-years. J Cyst Fibros 14(3): 361-369.
- Smith WD, Bardin E, Cameron L, Edmondson CL, Farrant KV, et al. (2017) Current and future therapies for Pseudomonas aeruginosa infection in patients with cystic fibrosis. FEMS microbiology letters 364(14).
- Akkerman-Nijland AM, Yousofi M, Rottier BL, Van der Vaart H, Burgerhof J, et al. (2020) Eradication of Pseudomonas aeruginosa in cystic fibrosis patients with inhalation of dry powder tobramycin. Ther Adv Respir Dis 14: 1753466620905279.
- Garcia-Clemente M, David de la Rosa D, Máiz L, Girón RB (2020) Impact of Pseudomonas aeruginosa Infection on Patients with Chronic Inflammatory Airway Diseases. J Clinical Med 9(12): 3800.
- Pang Z, Raudonis R, Glick BR, Lin TJ, Cheng Z (2019) Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol Adv 37(1): 177-192.
- Anghel I, Grumezescu A, Holban A, Gheorghe I, Vlad M, et al. (2013) Improved activity of aminoglycosides entrapped in silica networks against microbial strains isolated from otolaryngological infections. Farmacia 62: 69-78.
- Garazzino S, Altieri E, Silvestro E, Pruccoli G, Scolfaro, et al. (2020) Ceftolozane/Tazobactam for Treating Children with Exacerbations of Cystic Fibrosis Due to Pseudomonas aeruginosa. Front Pediatr 8: 173.
- Ottino L, Bartalesi F, Borchi B, Bresci S, Cavallo A, et al. (2021) ceftolozane/tazobactam for Pseudomonas aeruginosa pulmonary exacerbations in cystic fibrosis adult patients: a case series. Eur J Clin Microbiol Infect Dis 40(10): 2211-2215.
- Kimberle CC, Patrick RM (2003) Principles of stains and media. In manual of clinical microbiology. Patrick RM, Ellen JB, James HJ, Michael AP, Robert HY, ASM PRESS, USA.
- Dumitru B (1999) Diagnosticul de laborator al infectiilor tractusului respirator inferior. In Tratat de Microbiologie Clinica. Dumitru Buiuc, Marian Negut, Editura Medicala, Romania.
- (2021) Hemocultura aeroba (cu antibiograma dupa caz). Synevo.
- Joyanes P, del Carmen Conejo M, Martínez-Martínez L, Perea, EJ (2001) Evaluation of the VITEK 2 system for the identification and susceptibility testing of three species of nonfermenting gram-negative rods frequently isolated from clinical samples. J Clinical Microbiol 39(9): 3247-3253.
- Abdul WA, Zahraldin K, Sid Ahmed MA, Jarir SA, Muneer M, et al. (2017) The emergence of multidrug-resistant Pseudomonas aeruginosa in cystic fibrosis patients on inhaled antibiotics. Lung India: 34(6): 527-531.
- Ciuca IM, Pop LL, Rogobete AF, Onet DI, Guta-Almajan B, et al. (2016) Genetic expression in cystic fibrosis related bone disease: An observational, transversal, cross-sectional study. Clinical laboratory 62(9): 1725-1730.
- Granados A, Chan CL, Ode KL, Moheet A, Moran A (2019) Cystic fibrosis related diabetes: Pathophysiology, screening and diagnosis. Journal of cystic fibrosis. J Cyst Fibros 18 Suppl 2: S3-S9.
- Prentice BJ, Jaffe A, Hameed S, Verge CF, Waters S, et al. (2021) Cystic fibrosis-related diabetes and lung disease: an update. Journal of the European Respiratory Society 30(159): 200293.
- Dediu M, Ciuca IM, Marc MS, Boeriu E, Pop LL (2021) Factors influencing lung function in patients with cystic fibrosis in western Romania. Journal of multidisciplinary healthcare 14: 1423-1429.
- Abdelbary S (2020) Recent approach in microbial pathogen complications in patients with cystic fibrosis. Cystic Fibrosis.
- Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, et al. (2012) Multidrug-resistant, extensively drug-resistant and pan drugresistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 18(3): 268-281.
- Lucca F, Guarnieri M, Ros M, Muffato G, Rigoli R, et al. (2018) Antibiotic resistance evolution of Pseudomonas aeruginosa in cystic fibrosis patients (2010-2013). Clinical Respir J 12(7): 2189-2196.
- Rutter WC, Burgess DR, Burgess DS (2017) Increasing Incidence of Multidrug Resistance Among Cystic Fibrosis Respiratory Bacterial Isolates. Microbial Drug Resis 23(1): 51-55.
- Ideozu JE, Zhang X, Pan A, Ashrafi Z, Woods KJ, et al. (2017) Increased Expression of Plasma-Induced ABCC1 mRNA in Cystic Fibrosis. International journal of molecular sciences 18(8): 1752.
- Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, et al. (2001) Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. Journal Infecti Dis 183(3): 444-452.
- Kosorok MR, Zeng L, Wes SE, Rock MJ, Splaingard, et al. (2001) Acceleration of lung disease in children with cystic fibrosis after Pseudomonas aeruginosa acquisition. Pediatr Pulmonol 32(4): 277-287.
- Li Z, Kosorok MR, Farrell PM, Laxova A, West SE, et al. (2005) Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 293(5): 581- 588.
- Moreau-Marquis S, Bomberger JM, Anderson GG, Swiatecka-Urban A, Ye S, et al. (2008) The deltaF508-CFTR mutation results in increased biofilm formation by Pseudomonas aeruginosa by increasing iron availability. American journal of physiology. Am J Physiol Lung Cell Mol Physiol 295(1): L25-L37.
- Ekkelenkamp MB, Cantón R, Díez-Aguilar M, Tunney MM, Gilpin, et al. (2020) Susceptibility of Pseudomonas aeruginosa recovered from cystic fibrosis patients to murepavadin and 13 comparator antibiotics. Antimicrobial agents and chemotherapy 64(2): e01541-e01519.
- Mustafa MH, Chalhoub H, Denis O, Deplano A, Vergison A, et al. (2016) Antimicrobial susceptibility of Pseudomonas aeruginosa isolated from cystic fibrosis patients in northern Europe. Antimicrob Agents Chemother 60(11): 6735-6741.
- Jansen G, Mahrt N, Tueffers L, Barbosa C, Harjes M, et al. (2016) Association between clinical antibiotic resistance and susceptibility of Pseudomonas in the cystic fibrosis lung. Evol Med Public Health 2016(1): 182-194.
- Çopur ÇA, Ertürk A, Ejder N, Rakici E, Kostakoğlu U, et al. (2021) screening of antimicrobial resistance genes and epidemiological features in hospital and community-associated carbapenem-resistant Pseudomonas aeruginosa infections. Infection and drug resistance 14: 1517-1526.
- (2020) European Centre for Disease Prevention and Control. Antimicrobial resistance in the EU/EEA (EARS-Net).
Article Type
Research Article
Publication history
Received Date: December 17, 2021
Published: January 11, 2022
Address for correspondence
Elena Camelia Berghea, Department of Pediatrics, Carol Davila University of Medicine and Pharmacy, Romania
Copyright
©2022 Open Access Journal of Biomedical Science, All rights reserved. No part of this content may be reproduced or transmitted in any form or by any means as per the standard guidelines of fair use. Open Access Journal of Biomedical Science is licensed under a Creative Commons Attribution 4.0 International License
How to cite this article
Marcela DI, Mihaela B, Maria CS, Maria IB, Andrei C, etc. Antibiotic Resistance of Pseudomonas aeruginosa Chronic Infection in Cystic Fibrosis Patients. A Single- Center Long-Term Follow-Up. 2022- 4(1) OAJBS.ID.000376.
Figure 1: Study design.

Figure 2: Change in antibiotic sensitivity of Pseudomonas aeruginosa.
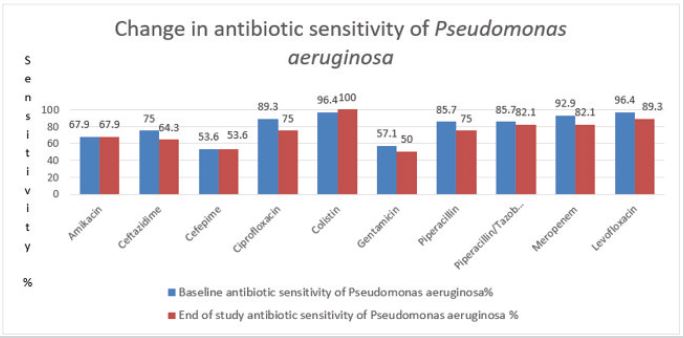
Table 1: Cohort characteristics.
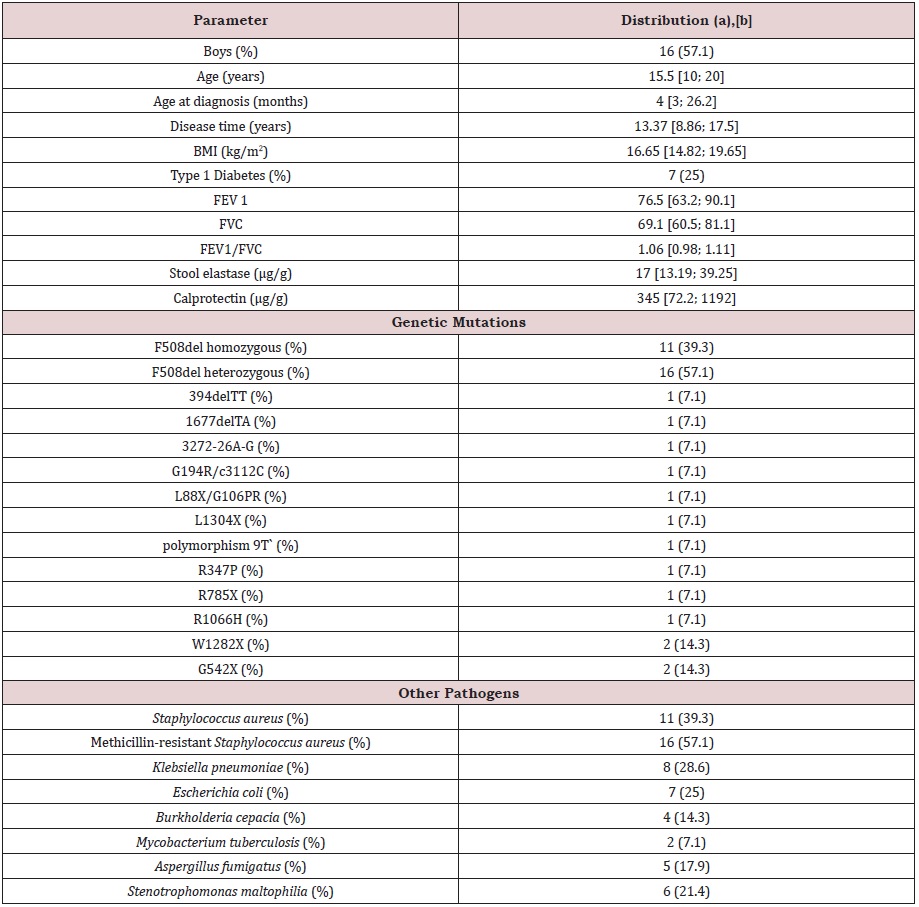
(a) Categorical variables were presented as numbers and in round brackets as percentages.
[b] Continuous variables were presented as the median and in square brackets as the interval between the quartiles.
Table 2: Antimicrobial Susceptibility of Pseudomonas aeruginosa.
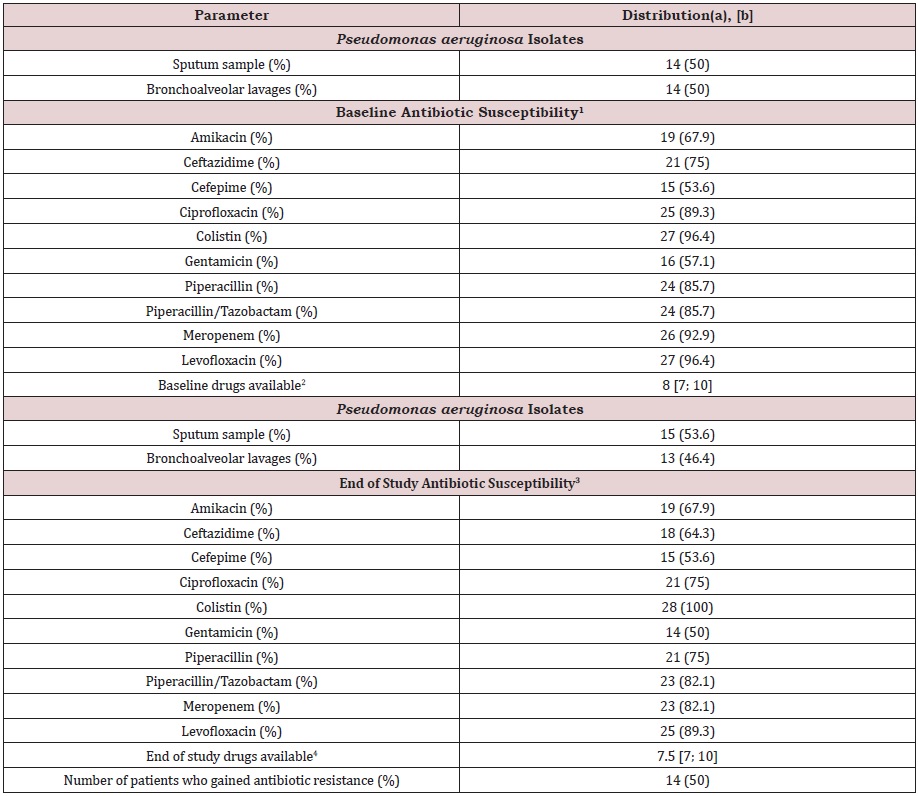
1The antibiotic sensitivity at the beginning of the study.
2The number of antibiotics that are sensitive to Pseudomonas aeruginosa infection at the beginning of the study.
3The antibiotic sensitivity at the end of the study.
4The number of antibiotics which are sensitive to Pseudomonas aeruginosa infection at the end of the study.
(a) Categorical variables were presented as numbers and in round brackets as percentages.
[b] Continuous variables were presented as the median and in square brackets as the interval between the quartiles.
